#4966 closed enhancement (fixed)
Alphafold: fetch the predicted aligned error file
| Reported by: | Tristan Croll | Owned by: | Tom Goddard |
|---|---|---|---|
| Priority: | normal | Milestone: | |
| Component: | Input/Output | Version: | |
| Keywords: | Cc: | ||
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
The following bug report has been submitted:
Platform: Linux-3.10.0-1160.25.1.el7.x86_64-x86_64-with-glibc2.17
ChimeraX Version: 1.3.dev202107240347 (2021-07-24 03:47:26 UTC)
Description
Really loving the new alphafold plugin! Just wondering... would you be willing to add a small convenience function to fetch the predicted aligned error file (has the format AF-{UniProt ID}-F1-predicted_aligned_error_v1.json) via the same API? This probably won't be useful for visualisation, but provides really useful information for weighting distance restraints (it's a 2D matrix giving the predicted error in CA-CA distance for every pair of residues in the model).
OpenGL version: 3.3.0 NVIDIA 465.19.01
OpenGL renderer: NVIDIA TITAN Xp/PCIe/SSE2
OpenGL vendor: NVIDIA Corporation
Manufacturer: Dell Inc.
Model: Precision T5600
OS: CentOS Linux 7 Core
Architecture: 64bit ELF
Virutal Machine: none
CPU: 32 Intel(R) Xeon(R) CPU E5-2687W 0 @ 3.10GHz
Cache Size: 20480 KB
Memory:
total used free shared buff/cache available
Mem: 62G 23G 22G 320M 16G 38G
Swap: 4.9G 0B 4.9G
Graphics:
03:00.0 VGA compatible controller [0300]: NVIDIA Corporation GP102 [TITAN Xp] [10de:1b02] (rev a1)
Subsystem: NVIDIA Corporation Device [10de:11df]
Kernel driver in use: nvidia
Locale: ('en_GB', 'UTF-8')
PyQt5 5.15.2, Qt 5.15.2
Installed Packages:
alabaster: 0.7.12
appdirs: 1.4.4
Babel: 2.9.1
backcall: 0.2.0
blockdiag: 2.0.1
certifi: 2021.5.30
cftime: 1.5.0
chardet: 4.0.0
ChimeraX-AddCharge: 1.1.4
ChimeraX-AddH: 2.1.10
ChimeraX-AlignmentAlgorithms: 2.0
ChimeraX-AlignmentHdrs: 3.2
ChimeraX-AlignmentMatrices: 2.0
ChimeraX-Alignments: 2.1
ChimeraX-AlphaFold: 1.0
ChimeraX-AltlocExplorer: 1.0
ChimeraX-AmberInfo: 1.0
ChimeraX-Arrays: 1.0
ChimeraX-Atomic: 1.27.1
ChimeraX-AtomicLibrary: 4.0
ChimeraX-AtomSearch: 2.0
ChimeraX-AtomSearchLibrary: 1.0
ChimeraX-AxesPlanes: 2.0
ChimeraX-BasicActions: 1.1
ChimeraX-BILD: 1.0
ChimeraX-BlastProtein: 1.1.1
ChimeraX-BondRot: 2.0
ChimeraX-BugReporter: 1.0
ChimeraX-BuildStructure: 2.5.2
ChimeraX-Bumps: 1.0
ChimeraX-BundleBuilder: 1.1
ChimeraX-ButtonPanel: 1.0
ChimeraX-CageBuilder: 1.0
ChimeraX-CellPack: 1.0
ChimeraX-Centroids: 1.1
ChimeraX-ChemGroup: 2.0
ChimeraX-Clashes: 2.1
ChimeraX-Clipper: 0.17.0
ChimeraX-ColorActions: 1.0
ChimeraX-ColorGlobe: 1.0
ChimeraX-ColorKey: 1.3.2
ChimeraX-CommandLine: 1.1.4
ChimeraX-ConnectStructure: 2.0
ChimeraX-Contacts: 1.0
ChimeraX-Core: 1.3.dev202107240347
ChimeraX-CoreFormats: 1.0
ChimeraX-coulombic: 1.3
ChimeraX-Crosslinks: 1.0
ChimeraX-Crystal: 1.0
ChimeraX-CrystalContacts: 1.0
ChimeraX-DataFormats: 1.2
ChimeraX-Dicom: 1.0
ChimeraX-DistMonitor: 1.1.4
ChimeraX-DistUI: 1.0
ChimeraX-Dssp: 2.0
ChimeraX-EMDB-SFF: 1.0
ChimeraX-ExperimentalCommands: 1.0
ChimeraX-FileHistory: 1.0
ChimeraX-FunctionKey: 1.0
ChimeraX-Geometry: 1.1
ChimeraX-gltf: 1.0
ChimeraX-Graphics: 1.1
ChimeraX-Hbonds: 2.1
ChimeraX-Help: 1.1
ChimeraX-HKCage: 1.3
ChimeraX-IHM: 1.1
ChimeraX-ImageFormats: 1.1
ChimeraX-IMOD: 1.0
ChimeraX-IO: 1.0.1
ChimeraX-ISOLDE: 1.3.dev32
ChimeraX-ItemsInspection: 1.0
ChimeraX-Label: 1.1
ChimeraX-LinuxSupport: 1.0
ChimeraX-ListInfo: 1.1.1
ChimeraX-Log: 1.1.4
ChimeraX-LookingGlass: 1.1
ChimeraX-Maestro: 1.8.1
ChimeraX-Map: 1.1
ChimeraX-MapData: 2.0
ChimeraX-MapEraser: 1.0
ChimeraX-MapFilter: 2.0
ChimeraX-MapFit: 2.0
ChimeraX-MapSeries: 2.1
ChimeraX-Markers: 1.0
ChimeraX-Mask: 1.0
ChimeraX-MatchMaker: 1.2.1
ChimeraX-MDcrds: 2.4
ChimeraX-MedicalToolbar: 1.0.1
ChimeraX-Meeting: 1.0
ChimeraX-MLP: 1.1
ChimeraX-mmCIF: 2.3
ChimeraX-MMTF: 2.1
ChimeraX-Modeller: 1.0.2
ChimeraX-ModelPanel: 1.1
ChimeraX-ModelSeries: 1.0
ChimeraX-Mol2: 2.0
ChimeraX-Morph: 1.0
ChimeraX-MouseModes: 1.1
ChimeraX-Movie: 1.0
ChimeraX-Neuron: 1.0
ChimeraX-Nucleotides: 2.0.2
ChimeraX-OpenCommand: 1.6.2
ChimeraX-PDB: 2.4.4
ChimeraX-PDBBio: 1.0
ChimeraX-PDBLibrary: 1.0.1
ChimeraX-PDBMatrices: 1.0
ChimeraX-Phenix: 0.3
ChimeraX-PickBlobs: 1.0
ChimeraX-Positions: 1.0
ChimeraX-PresetMgr: 1.0.1
ChimeraX-PubChem: 2.1
ChimeraX-ReadPbonds: 1.0
ChimeraX-Registration: 1.1
ChimeraX-RemoteControl: 1.0
ChimeraX-ResidueFit: 1.0
ChimeraX-RestServer: 1.1
ChimeraX-RNALayout: 1.0
ChimeraX-RotamerLibMgr: 2.0
ChimeraX-RotamerLibsDunbrack: 2.0
ChimeraX-RotamerLibsDynameomics: 2.0
ChimeraX-RotamerLibsRichardson: 2.0
ChimeraX-Sample: 0.1
ChimeraX-SaveCommand: 1.4.1
ChimeraX-SchemeMgr: 1.0
ChimeraX-SDF: 2.0
ChimeraX-Segger: 1.0
ChimeraX-Segment: 1.0
ChimeraX-SelInspector: 1.0
ChimeraX-SeqView: 2.4.1
ChimeraX-Shape: 1.0.1
ChimeraX-Shell: 1.0
ChimeraX-Shortcuts: 1.1
ChimeraX-ShowAttr: 1.0
ChimeraX-ShowSequences: 1.0
ChimeraX-SideView: 1.0
ChimeraX-Smiles: 2.1
ChimeraX-SmoothLines: 1.0
ChimeraX-SpaceNavigator: 1.0
ChimeraX-StdCommands: 1.6
ChimeraX-STL: 1.0
ChimeraX-Storm: 1.0
ChimeraX-Struts: 1.0
ChimeraX-Surface: 1.0
ChimeraX-SwapAA: 2.0
ChimeraX-SwapRes: 2.1
ChimeraX-TapeMeasure: 1.0
ChimeraX-Test: 1.0
ChimeraX-Toolbar: 1.1
ChimeraX-ToolshedUtils: 1.2
ChimeraX-Tug: 1.0
ChimeraX-UI: 1.10.1
ChimeraX-uniprot: 2.1
ChimeraX-UnitCell: 1.0
ChimeraX-ViewDockX: 1.0.1
ChimeraX-Vive: 1.1
ChimeraX-VolumeMenu: 1.0
ChimeraX-Voyager: 0.1
ChimeraX-VTK: 1.0
ChimeraX-WavefrontOBJ: 1.0
ChimeraX-WebCam: 1.0
ChimeraX-WebServices: 1.0
ChimeraX-Zone: 1.0
colorama: 0.4.4
comtypes: 1.1.10
cxservices: 1.0
cycler: 0.10.0
Cython: 0.29.23
decorator: 4.4.2
distlib: 0.3.1
distro: 1.5.0
docutils: 0.17.1
filelock: 3.0.12
funcparserlib: 0.3.6
grako: 3.16.5
h5py: 3.3.0
html2text: 2020.1.16
idna: 2.10
ihm: 0.20
imagecodecs: 2021.4.28
imagesize: 1.2.0
ipykernel: 5.5.5
ipython: 7.23.1
ipython-genutils: 0.2.0
jedi: 0.18.0
Jinja2: 2.11.3
jupyter-client: 6.1.12
jupyter-core: 4.7.1
kiwisolver: 1.3.1
line-profiler: 3.3.0
lxml: 4.6.3
lz4: 3.1.3
MarkupSafe: 1.1.1
matplotlib: 3.4.2
matplotlib-inline: 0.1.2
msgpack: 1.0.2
netCDF4: 1.5.6
networkx: 2.5.1
numexpr: 2.7.3
numpy: 1.21.0
numpydoc: 1.1.0
OpenMM: 7.6.0
openvr: 1.16.801
packaging: 21.0
ParmEd: 3.2.0
parso: 0.8.2
pexpect: 4.8.0
pickleshare: 0.7.5
Pillow: 8.2.0
pip: 21.1.1
pkginfo: 1.7.0
prompt-toolkit: 3.0.19
psutil: 5.8.0
ptyprocess: 0.7.0
pycollada: 0.7.1
pydicom: 2.1.2
Pygments: 2.9.0
PyOpenGL: 3.1.5
PyOpenGL-accelerate: 3.1.5
pyparsing: 2.4.7
PyQt5-commercial: 5.15.2
PyQt5-sip: 12.8.1
PyQtWebEngine-commercial: 5.15.2
python-dateutil: 2.8.1
pytz: 2021.1
pyzmq: 22.1.0
qtconsole: 5.1.0
QtPy: 1.9.0
RandomWords: 0.3.0
requests: 2.25.1
scipy: 1.6.3
setuptools: 57.0.0
sfftk-rw: 0.7.0.post1
six: 1.16.0
snowballstemmer: 2.1.0
sortedcontainers: 2.4.0
Sphinx: 4.0.1
sphinxcontrib-applehelp: 1.0.2
sphinxcontrib-blockdiag: 2.0.0
sphinxcontrib-devhelp: 1.0.2
sphinxcontrib-htmlhelp: 2.0.0
sphinxcontrib-jsmath: 1.0.1
sphinxcontrib-qthelp: 1.0.3
sphinxcontrib-serializinghtml: 1.1.5
suds-jurko: 0.6
tables: 3.6.1
tifffile: 2021.4.8
tinyarray: 1.2.3
tornado: 6.1
traitlets: 5.0.5
urllib3: 1.26.6
wcwidth: 0.2.5
webcolors: 1.11.1
wheel: 0.36.2
wheel-filename: 1.3.0
Attachments (3)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Change History (28)
comment:1 by , 5 years ago
| Component: | Unassigned → Input/Output |
|---|---|
| Owner: | set to |
| Platform: | → all |
| Project: | → ChimeraX |
| Status: | new → assigned |
| Summary: | ChimeraX bug report submission → Alphafold: fetch the predicted aligned error file |
| Type: | defect → enhancement |
comment:2 by , 5 years ago
follow-up: 3 comment:3 by , 5 years ago
Fair enough points. I was thinking that having it in the AlphaFold plugin would minimise code duplication since the framework is already there - but the way you describe it here I suppose there's not much code to duplicate!
Apart from my main aim of using it to better weight distance restraints, another use I can think of would be partitioning the prediction into distinct domains (that you could then overlay separately onto the relevant parts of the reference model). But that would be a bit of work, and probably quite hard to make fast.
________________________________
From: ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu>
Sent: 28 July 2021 18:04
Cc: goddard@cgl.ucsf.edu <goddard@cgl.ucsf.edu>; Tristan Croll <tic20@cam.ac.uk>
Subject: Re: [ChimeraX] #4966: Alphafold: fetch the predicted aligned error file
#4966: Alphafold: fetch the predicted aligned error file
------------------------------------+-------------------------
Reporter: Tristan Croll | Owner: Tom Goddard
Type: enhancement | Status: assigned
Priority: normal | Milestone:
Component: Input/Output | Version:
Resolution: | Keywords:
Blocked By: | Blocking:
Notify when closed: | Platform: all
Project: ChimeraX |
------------------------------------+-------------------------
Comment (by Tom Goddard):
Wondering how you want to use this. Shouldn't the code that wants to use
it, for example in ISOLDE to generate restraints fetch the file? The
uniprot ids can be obtained from the new chimerax.atomic uniprot_ids()
method which is what the alphafold fetch uses to read uniprot ids from
mmCIF and PDB file headers. And the chimerax.core.fetch retrieve_url()
routine can fetch the file, or if you want it cached locally the
fetch_file() routine.
--
Ticket URL: <https://www.rbvi.ucsf.edu/trac/ChimeraX/ticket/4966#comment:2>
ChimeraX <https://www.rbvi.ucsf.edu/chimerax/>
ChimeraX Issue Tracker
follow-up: 4 comment:4 by , 5 years ago
Basic working implementation (fetching the PAE matrix and using it to partition a model into domains) at https://github.com/tristanic/isolde/tree/master/isolde/src/reference_model/alphafold. Requires NetworkX to be updated to 2.6.2 (adds a "resolution" keyword to the greedy_modularity_communities() method controlling the stringency of the clustering. Attached example for the insulin receptor (P06213) using the default arguments. Actually pretty quick, all told (a few seconds).
________________________________
From: ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu>
Sent: 28 July 2021 18:10
To: goddard@cgl.ucsf.edu <goddard@cgl.ucsf.edu>; Tristan Croll <tic20@cam.ac.uk>
Subject: Re: [ChimeraX] #4966: Alphafold: fetch the predicted aligned error file
#4966: Alphafold: fetch the predicted aligned error file
------------------------------------+-------------------------
Reporter: Tristan Croll | Owner: Tom Goddard
Type: enhancement | Status: assigned
Priority: normal | Milestone:
Component: Input/Output | Version:
Resolution: | Keywords:
Blocked By: | Blocking:
Notify when closed: | Platform: all
Project: ChimeraX |
------------------------------------+-------------------------
Comment (by Tristan Croll):
{{{
Fair enough points. I was thinking that having it in the AlphaFold plugin
would minimise code duplication since the framework is already there - but
the way you describe it here I suppose there's not much code to duplicate!
Apart from my main aim of using it to better weight distance restraints,
another use I can think of would be partitioning the prediction into
distinct domains (that you could then overlay separately onto the relevant
parts of the reference model). But that would be a bit of work, and
probably quite hard to make fast.
________________________________
From: ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu>
Sent: 28 July 2021 18:04
Cc: goddard@cgl.ucsf.edu <goddard@cgl.ucsf.edu>; Tristan Croll
<tic20@cam.ac.uk>
Subject: Re: [ChimeraX] #4966: Alphafold: fetch the predicted aligned
error file
#4966: Alphafold: fetch the predicted aligned error file
------------------------------------+-------------------------
Reporter: Tristan Croll | Owner: Tom Goddard
Type: enhancement | Status: assigned
Priority: normal | Milestone:
Component: Input/Output | Version:
Resolution: | Keywords:
Blocked By: | Blocking:
Notify when closed: | Platform: all
Project: ChimeraX |
------------------------------------+-------------------------
Comment (by Tom Goddard):
Wondering how you want to use this. Shouldn't the code that wants to use
it, for example in ISOLDE to generate restraints fetch the file? The
uniprot ids can be obtained from the new chimerax.atomic uniprot_ids()
method which is what the alphafold fetch uses to read uniprot ids from
mmCIF and PDB file headers. And the chimerax.core.fetch retrieve_url()
routine can fetch the file, or if you want it cached locally the
fetch_file() routine.
--
Ticket URL:
<https://www.rbvi.ucsf.edu/trac/ChimeraX/ticket/4966#comment:2>
ChimeraX <https://www.rbvi.ucsf.edu/chimerax/>
ChimeraX Issue Tracker
}}}
--
Ticket URL: <https://www.rbvi.ucsf.edu/trac/ChimeraX/ticket/4966#comment:3>
ChimeraX <https://www.rbvi.ucsf.edu/chimerax/>
ChimeraX Issue Tracker
follow-up: 5 comment:6 by , 5 years ago
| Resolution: | → fixed |
|---|---|
| Status: | assigned → closed |
Cool. Seems to pick up several fragments (e.g. loops) that also are not well connected and so become separate domains. Some kind of domain identification would be nice to have in ChimeraX.
I think we may have an alphafold command with various subcommands to for instance fetch a model, run a structure prediction, color in various ways. This domain identification could be added to that. How will these domains be used? Probably need something beyond coloring, like naming them. And probably a user will want to fix the mistakes, joining the fragments to the domain they belong to.
A related ticket on improvements to fetching AlphaFold models is #4969.
comment:7 by , 5 years ago
This isn’t true “domain identification” as such - it knows nothing about covalent connectivity, for a start. Rather, it’s picking out the blocks that AlphaFold2 predicts will be approximately rigid. I can think of a few applications for this - for some it would make sense to roll in the loops, but for others they’re best left out: - molecular replacement relies heavily on finding the biggest fragment(s) that will overlay rigidly with the structure in the crystal itself (which of course you don’t know ahead of time in most cases). Current methods tend to rely on manual identification of domains, then pruning by B-factor and truncating non-identical sidechains. Where there are multiple existing homologues it’s possible to do better by pruning to the parts that rigidly overlay each other, but there’s still room for improvement. Looks like the AF2 models could dramatically simplify that - decompose into domains based on predicted aligned error, then further prune based on predicted LDDT. - in a similar way, this could help people docking into cryo-EM density. Dock the individual rigid components then build in the connections - or perhaps even better, use the docking results to set targets for ISOLDE to flexibly fit the full chain. - for your existing “fetch AF2 models and overlay on the experimental structure” tool, this could let you identify big conformational differences and provide separate overlays for the different components. For example in 6o85, chains A and B each have a C-terminal domain connected by a long flexible linker - the AF2 model aligns to the N-terminus leaving the C-terminus in completely the wrong place.
follow-up: 7 comment:8 by , 5 years ago
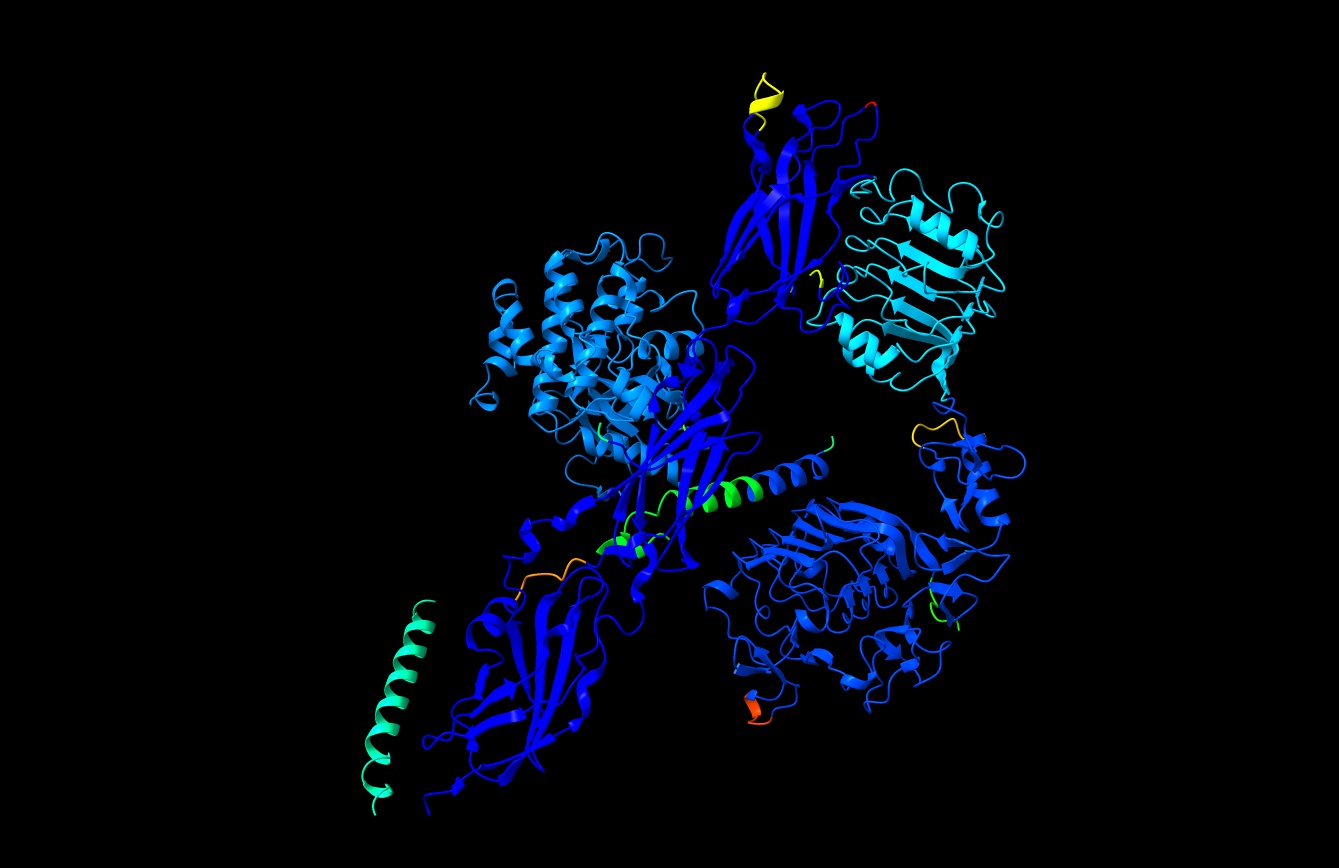
Case in point: in the image I attached of the insulin receptor model, that enormous dark cyan domain on the upper left is actually the kinase domain (and the bright-cyan helix on lower left is the transmembrane domain) - since AF2 knows nothing about membranes it's done some pretty weird gymnastics with these.
________________________________
From: ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu>
Sent: 30 July 2021 08:56
To: goddard@cgl.ucsf.edu <goddard@cgl.ucsf.edu>; Tristan Croll <tic20@cam.ac.uk>
Subject: Re: [ChimeraX] #4966: Alphafold: fetch the predicted aligned error file
#4966: Alphafold: fetch the predicted aligned error file
------------------------------------+-------------------------
Reporter: Tristan Croll | Owner: Tom Goddard
Type: enhancement | Status: closed
Priority: normal | Milestone:
Component: Input/Output | Version:
Resolution: fixed | Keywords:
Blocked By: | Blocking:
Notify when closed: | Platform: all
Project: ChimeraX |
------------------------------------+-------------------------
Comment (by Tristan Croll):
{{{
This isn’t true “domain identification” as such - it knows nothing about
covalent connectivity, for a start. Rather, it’s picking out the blocks
that AlphaFold2 predicts will be approximately rigid. I can think of a few
applications for this - for some it would make sense to roll in the loops,
but for others they’re best left out:
- molecular replacement relies heavily on finding the biggest fragment(s)
that will overlay rigidly with the structure in the crystal itself (which
of course you don’t know ahead of time in most cases). Current methods
tend to rely on manual identification of domains, then pruning by B-factor
and truncating non-identical sidechains. Where there are multiple existing
homologues it’s possible to do better by pruning to the parts that rigidly
overlay each other, but there’s still room for improvement. Looks like the
AF2 models could dramatically simplify that - decompose into domains based
on predicted aligned error, then further prune based on predicted LDDT.
- in a similar way, this could help people docking into cryo-EM density.
Dock the individual rigid components then build in the connections - or
perhaps even better, use the docking results to set targets for ISOLDE to
flexibly fit the full chain.
- for your existing “fetch AF2 models and overlay on the experimental
structure” tool, this could let you identify big conformational
differences and provide separate overlays for the different components.
For example in 6o85, chains A and B each have a C-terminal domain
connected by a long flexible linker - the AF2 model aligns to the
N-terminus leaving the C-terminus in completely the wrong place.
}}}
--
Ticket URL: <https://www.rbvi.ucsf.edu/trac/ChimeraX/ticket/4966#comment:7>
ChimeraX <https://www.rbvi.ucsf.edu/chimerax/>
ChimeraX Issue Tracker
follow-up: 8 comment:9 by , 5 years ago
Yes I see it could be useful for fitting rigid pieces or x-ray phasing searches.
comment:10 by , 5 years ago
Today I tried out using python-igraph in place of NetworkX for the community clustering. Gives very similar results but is much, much faster - roughly 40X overall, if the PAE matrix has already been imported to a numpy array and adjust_weights_for_distance=False (this was an option I wanted to try out, but doesn't seem to add much in general). Can get an extra ~30% speedup if pae_cutoff is reduced from 5 to 3. Overall runtime comes down from 6-10 seconds to ~150-250ms for my two test cases (847 and ~2450 residues). Fast enough to potentially become an interactive tool with sliders for the settings.
comment:11 by , 5 years ago
Would probably need python-igraph to be bundled within ChimeraX, though. For some reason, despite being distributed as Linux, Windows and MacOS wheels it still does some compilation on pip install (and fails in my ~vanilla CentOS 7 environment because it requires cmake 3).
comment:12 by , 5 years ago
Made some more intelligent use of Numpy for the CA-CA distance matrix, bringing the impact of adjust_weights_for_distance=True down from ~2s to ~70ms.
comment:13 by , 5 years ago
That is cool that igraph works well. But that is a nightmare that they don't allow you to install without build tools. Are you sure that is right? I have never seen a case where a binary wheel needs builds tools like cmake. Maybe it is that igraph has a dependency that is only available as a source wheel and needs cmake. I think we don't have cmake in our build environment and have rejected other packages that would need it to build. You know my position from OpenMM -- I am against including packages where the developers of those packages don't have the time to make an easy to use installation. We can't afford to do the work for them to make their packages usable.
comment:14 by , 5 years ago
Hmm... my bad. The pip install failure was on my system python 3.7 - turns out the wheel file required a newer version of pip - but rather than tell me that, it was just falling back to grabbing the source tarball. The binary wheel installs just fine in ChimeraX.
comment:15 by , 5 years ago
Great! So you can add the igraph dependency to your bundle. When we need graph manipulation for other tools we will definitely look at it as an alternative to networkx.
comment:16 by , 5 years ago
The funny thing is that I don't actually have an immediate use for this domain parsing in ISOLDE itself (although it might come in handy later as part of a toolkit for building up models into maps from scratch). But Airlie McCoy and Claudia Nebot in the lab here are very keen to set up a ChimeraX front-end to Phaser TNG (the new, heavily rewritten and re-thought version of Phaser: https://journals.iucr.org/d/issues/2021/01/00/ba5309/index.html). Still very early days on that one, but this will have clear applications there as a tool to generate suitable molecular replacement search models from a predicted structure.
________________________________
From: ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu>
Sent: 31 August 2021 17:35
Cc: goddard@cgl.ucsf.edu <goddard@cgl.ucsf.edu>; Tristan Croll <tic20@cam.ac.uk>
Subject: Re: [ChimeraX] #4966: Alphafold: fetch the predicted aligned error file
#4966: Alphafold: fetch the predicted aligned error file
------------------------------------+-------------------------
Reporter: Tristan Croll | Owner: Tom Goddard
Type: enhancement | Status: closed
Priority: normal | Milestone:
Component: Input/Output | Version:
Resolution: fixed | Keywords:
Blocked By: | Blocking:
Notify when closed: | Platform: all
Project: ChimeraX |
------------------------------------+-------------------------
Comment (by Tom Goddard):
Great! So you can add the igraph dependency to your bundle. When we need
graph manipulation for other tools we will definitely look at it as an
alternative to networkx.
--
Ticket URL: <https://www.rbvi.ucsf.edu/trac/ChimeraX/ticket/4966#comment:15>
ChimeraX <https://www.rbvi.ucsf.edu/chimerax/>
ChimeraX Issue Tracker
follow-up: 16 comment:17 by , 5 years ago
Ok. For cryoEM maps it could be useful to move domains to match the map to get an initial model. How to select the domains becomes a basic issue.
comment:18 by , 5 years ago
The way I'm doing it now is to define a new per-residue integer property (currently Residue.isolde_domain) and give all residues in a given cluster the same isolde_domain value. Makes a good handle for selecting the domains later. The general idea I'm thinking of would be to split the domains to be used for rigid docking into separate models, dock them, and then use the docked positions as targets to steer the (restrained with local distance and torsion restraints) complete chain in a simulation, allowing the connecting regions to adjust to suit.
Fast global docking into cryo-EM maps (and improved docking into difficult cryo-EM density) is one of the goals of the new Phaser, by the way.
comment:19 by , 5 years ago
That sounds like a reasonable plan. I was thinking of using the split command to make separate domain models, then doing rigid fitting with the fitmap command, the copying the coordinates from the domains back to the unsplit structure using the mcopy command (which needs to be ported from Chimera to ChimeraX). This would just be done by the user by hand.
comment:20 by , 5 years ago
I wonder if the ChimeraX model morphing code could be used to help define a more meaningful steering trajectory than simple position restraints, for the cases where domains have to rotate substantially?
________________________________
From: ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu>
Sent: 31 August 2021 22:09
Cc: goddard@cgl.ucsf.edu <goddard@cgl.ucsf.edu>; Tristan Croll <tic20@cam.ac.uk>
Subject: Re: [ChimeraX] #4966: Alphafold: fetch the predicted aligned error file
#4966: Alphafold: fetch the predicted aligned error file
------------------------------------+-------------------------
Reporter: Tristan Croll | Owner: Tom Goddard
Type: enhancement | Status: closed
Priority: normal | Milestone:
Component: Input/Output | Version:
Resolution: fixed | Keywords:
Blocked By: | Blocking:
Notify when closed: | Platform: all
Project: ChimeraX |
------------------------------------+-------------------------
Comment (by Tom Goddard):
That sounds like a reasonable plan. I was thinking of using the split
command to make separate domain models, then doing rigid fitting with the
fitmap command, the copying the coordinates from the domains back to the
unsplit structure using the mcopy command (which needs to be ported from
Chimera to ChimeraX). This would just be done by the user by hand.
--
Ticket URL: <https://www.rbvi.ucsf.edu/trac/ChimeraX/ticket/4966#comment:19>
ChimeraX <https://www.rbvi.ucsf.edu/chimerax/>
ChimeraX Issue Tracker
follow-up: 20 comment:21 by , 5 years ago
Morphing is just driving torsion angles. Possibly you could use the rigid rotation computed by the align command and apply a torque to the whole domain to rotate it. But maybe not necessary and I would first want to see a case where just position restraints has trouble and there is a large rotation before working on that.
comment:22 by , 4 years ago
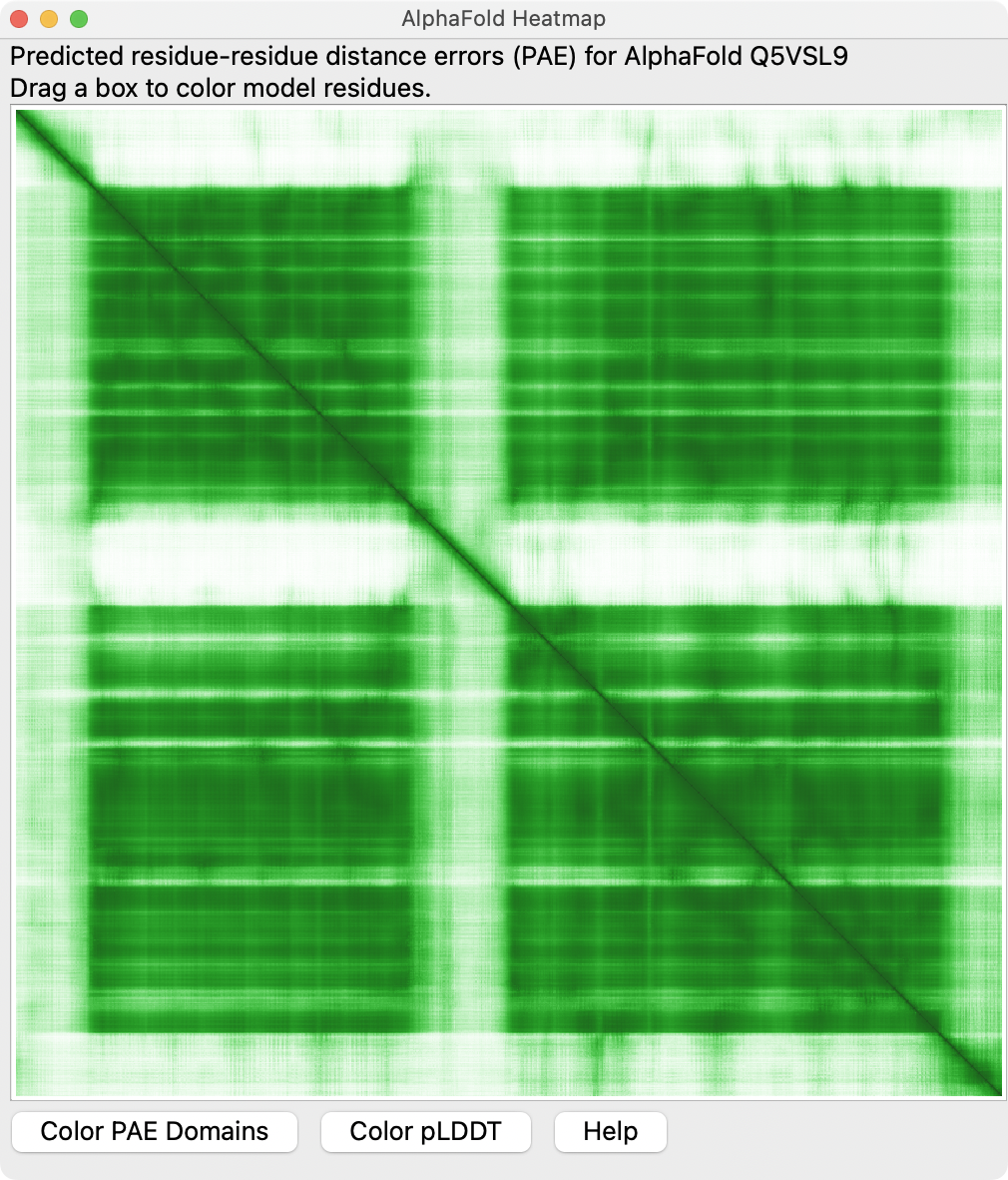
I added the ability to show AlphaFold predicted aligned error as a heat map, and to color residues by domains determined by Tristan's networkx cluster code. The heat map display allows dragging a box around any region on the heat map to color the corresponding residues. This is copied from the AlphaFold database web pages, including the green color scheme for the heat map. To fetch the PAE matrix data from the AlphaFold database there is a new "pae" option to the alphafold fetch command, for example
alphafold fetch q5vsl9 pae true
If instead you want to open the PAE JSON or pickle file from an AlphaFold run you can use the new command "alphafold pae", for instance,
alphafold pae result_model_1_multimer.pkl model #2
Here you specify which atomic structure model to associate with the PAE data. If only one atomic structure is open you can omit the model option.
Both these commands display the heat map panel which has buttons "Color PAE Domains" and "Color pLDDT". The first computes the domains and colors each a different (random) color. The second gives the standard AlphaFold blue-red per-residue pLDDT score coloring using the bfactor field of the structure.
I've attached an image of the new heat map panel.
by , 4 years ago
| Attachment: | heatmap.png added |
|---|
by , 4 years ago
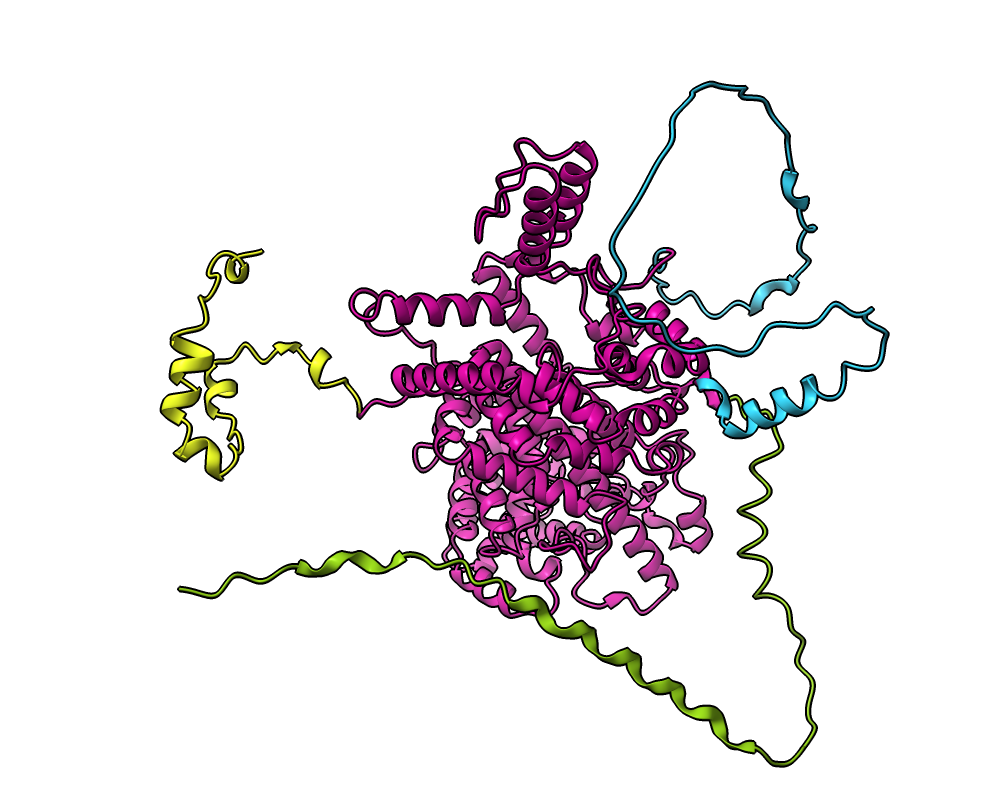
| Attachment: | q5vsl9_domains.png added |
|---|
Domain coloring for AlphaFold database entry Q5VSL9 showing one core domain, and 3 disordered regions.
comment:23 by , 4 years ago
I hope to show this PAE heatmap display in an AlphaFold SBGrid webinar talk I am giving on Tuesday March 29, 2022.
comment:24 by , 4 years ago
This is most excellent, and will be really useful! I'm particularly thankful that you've added the ability to load the PAE matrix from a user-generated prediction. A couple of suggestions:
- for the heatmap display I think it would be a nice touch to add a slider to adjust the maximum of the colormap range - while the PAE values go up to 32-ish, for many purposes the most interesting PAE values are below around 4A.
- for the domain parsing, it would be a good idea to expose the
resolutionargument of the community clustering method in some way (perhaps as a combo box with a step size of 0.5 or so). It controls how aggressive the algorithm is when defining individual clusters, and the optimum does tend to vary a bit from case to case (and application to application) - I haven't found a really good one-size-fits-all, but useful values are generally in the range of 0.5-5.
comment:25 by , 4 years ago
Thanks for the suggestions. I've added options to the "alphafold pae" command to set the range of PAE values for the colormap and also for the colors to use (range and palette options). And I added command options to do the domain coloring (option "color_domains") and specify graph resolution (option "clustering", default 0.5) and cutoff (option "connect_max_pae", default 5). I'll add options to the gui when it becomes clearer what options are most commonly useful. So far I did not find using more limited pae color range useful.
Wondering how you want to use this. Shouldn't the code that wants to use it, for example in ISOLDE to generate restraints fetch the file? The uniprot ids can be obtained from the new chimerax.atomic uniprot_ids() method which is what the alphafold fetch uses to read uniprot ids from mmCIF and PDB file headers. And the chimerax.core.fetch retrieve_url() routine can fetch the file, or if you want it cached locally the fetch_file() routine.