#8473 closed defect (fixed)
Show chain sequence: TypeError: __init__() got an unexpected keyword argument 'seq_viewer'
| Reported by: | Owned by: | Eric Pettersen | |
|---|---|---|---|
| Priority: | major | Milestone: | 1.6 |
| Component: | Sequence | Version: | |
| Keywords: | Cc: | ||
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
The following bug report has been submitted:
Platform: Windows-10-10.0.19044
ChimeraX Version: 1.6.dev202302100235 (2023-02-10 02:35:09 UTC)
Description
(Describe the actions that caused this problem to occur here)
The sequence viewer tool does not work properly anymore in the ChimeraX daily build (2023/02/09) version.
(By the way: I used the daily build, because ChimeraX 1.5 has a bug which is raised upon once using the "repaired" DockView tool with a SwissDock output: the "models" window now remains permanently / by default unchecked in the tools menu and the "models" tab permanently appears as separate window upon ChimeraX startup - which requires manuel window closing (or double-clicking in the side panel to place it there; it's really "annoying".)
Back on topic:
Did a matchmaker overlay with 3 models of the same protein (2 of these in complex with another protein: a homodimer model plus a heterodimer overlayed on the monomeric model) and wanted to get the same sequence regions highlighted in the viewing panel.
Just an error message appears with and/or without same sequences grouped/not grouped:
ui tool show "Show Sequence Viewer"
sequence chain #1/A #2/A #2/B #3/A
Alignment identifier is 1 Traceback (most recent call last): File "C:\Program Files\ChimeraX\bin\lib\site-packages\chimerax\show_sequences\tool.py", line 64, in show_seqs run(self.session, "seq chain %s" % " ".join([chain.atomspec for chain in chains])) File "C:\Program Files\ChimeraX\bin\lib\site-packages\chimerax\core\commands\run.py", line 38, in run results = command.run(text, log=log, return_json=return_json) File "C:\Program Files\ChimeraX\bin\lib\site-packages\chimerax\core\commands\cli.py", line 2897, in run result = ci.function(session, **kw_args) File "C:\Program Files\ChimeraX\bin\lib\site-packages\chimerax\seqalign\cmd.py", line 232, in seqalign_chain alignment = session.alignments.new_alignment([seq], None, seq_viewer="sv", File "C:\Program Files\ChimeraX\bin\lib\site-packages\chimerax\seqalign\manager.py", line 242, in new_alignment alignment = Alignment(self.session, seqs, identify_as, attrs, markups, auto_destroy, TypeError: __init__() got an unexpected keyword argument 'seq_viewer' TypeError: __init__() got an unexpected keyword argument 'seq_viewer' File "C:\Program Files\ChimeraX\bin\lib\site-packages\chimerax\seqalign\manager.py", line 242, in new_alignment alignment = Alignment(self.session, seqs, identify_as, attrs, markups, auto_destroy, See log for complete Python traceback.
ui tool show "Show Sequence Viewer"
sequence chain #1/A
Alignment identifier is 1/A Traceback (most recent call last): File "C:\Program Files\ChimeraX\bin\lib\site-packages\chimerax\show_sequences\tool.py", line 64, in show_seqs run(self.session, "seq chain %s" % " ".join([chain.atomspec for chain in chains])) File "C:\Program Files\ChimeraX\bin\lib\site-packages\chimerax\core\commands\run.py", line 38, in run results = command.run(text, log=log, return_json=return_json) File "C:\Program Files\ChimeraX\bin\lib\site-packages\chimerax\core\commands\cli.py", line 2897, in run result = ci.function(session, **kw_args) File "C:\Program Files\ChimeraX\bin\lib\site-packages\chimerax\seqalign\cmd.py", line 212, in seqalign_chain alignment = session.alignments.new_alignment([chain], ident, seq_viewer="sv", File "C:\Program Files\ChimeraX\bin\lib\site-packages\chimerax\seqalign\manager.py", line 242, in new_alignment alignment = Alignment(self.session, seqs, identify_as, attrs, markups, auto_destroy, TypeError: __init__() got an unexpected keyword argument 'seq_viewer' TypeError: __init__() got an unexpected keyword argument 'seq_viewer' File "C:\Program Files\ChimeraX\bin\lib\site-packages\chimerax\seqalign\manager.py", line 242, in new_alignment alignment = Alignment(self.session, seqs, identify_as, attrs, markups, auto_destroy, See log for complete Python traceback.
Log:
UCSF ChimeraX version: 1.6.dev202302100235 (2023-02-10)
© 2016-2023 Regents of the University of California. All rights reserved.
> open C:\\\MW\\\MW_2022\\\Arnim\\\antibody_project\\\CAD_CAD2_CADiCAD_Fab-
> full.cxs
Log from Fri Feb 10 17:20:21 2023 Startup Messages
---
warnings | QWindowsWindow::setDarkBorderToWindow: Unable to set dark window
border.
QWindowsWindow::setDarkBorderToWindow: Unable to set dark window border.
note | available bundle cache has not been initialized yet
UCSF ChimeraX version: 1.6.dev202302100235 (2023-02-10)
© 2016-2023 Regents of the University of California. All rights reserved.
How to cite UCSF ChimeraX
> open "C:/MW/MW_2022/Arnim/CAD Docking/proteins &
> ligands/2022-12-16/AF-O76075-F1-model_v2.pdb"
AF-O76075-F1-model_v2.pdb title:
Alphafold monomer V2.0 prediction for DNA fragmentation factor subunit β
(O76075) [more info...]
Chain information for AF-O76075-F1-model_v2.pdb #1
---
Chain | Description | UniProt
A | DNA fragmentation factor subunit β | DFFB_HUMAN 1-338
> lighting full
> open "C:/MW/MW_2022/Arnim/CAD Docking/proteins &
> ligands/CAD_dimer_Deepmind_selected_prediction.pdb"
Chain information for CAD_dimer_Deepmind_selected_prediction.pdb #2
---
Chain | Description
A B | No description available
> matchmaker #2 to #1
Parameters
---
Chain pairing | bb
Alignment algorithm | Needleman-Wunsch
Similarity matrix | BLOSUM-62
SS fraction | 0.3
Gap open (HH/SS/other) | 18/18/6
Gap extend | 1
SS matrix | | | H | S | O
---|---|---|---
H | 6 | -9 | -6
S | | 6 | -6
O | | | 4
Iteration cutoff | 2
Matchmaker AF-O76075-F1-model_v2.pdb, chain A (#1) with
CAD_dimer_Deepmind_selected_prediction.pdb, chain A (#2), sequence alignment
score = 1738.9
RMSD between 235 pruned atom pairs is 0.565 angstroms; (across all 338 pairs:
3.948)
> open "C:/MW/MW_2022/Arnim/CAD Docking/proteins &
> ligands/CAD_iCAD_dimer_Deepmind_selected_prediction.pdb"
Chain information for CAD_iCAD_dimer_Deepmind_selected_prediction.pdb #3
---
Chain | Description
A | No description available
B | No description available
> matchmaker #3 to #1
Parameters
---
Chain pairing | bb
Alignment algorithm | Needleman-Wunsch
Similarity matrix | BLOSUM-62
SS fraction | 0.3
Gap open (HH/SS/other) | 18/18/6
Gap extend | 1
SS matrix | | | H | S | O
---|---|---|---
H | 6 | -9 | -6
S | | 6 | -6
O | | | 4
Iteration cutoff | 2
Matchmaker AF-O76075-F1-model_v2.pdb, chain A (#1) with
CAD_iCAD_dimer_Deepmind_selected_prediction.pdb, chain A (#3), sequence
alignment score = 1713.1
RMSD between 79 pruned atom pairs is 0.270 angstroms; (across all 338 pairs:
34.008)
> hide #1 models
> show #1 models
> hide #3 models
> color #1-2 bychain
> hide #2 models
> show #2 models
> hide #2 models
> show #2 models
> hide #2 models
> color #1 #55aaffff
> show #2 models
> hide #2 models
> show #3 models
> hide #1 models
> show #2 models
> hide #2 models
> hide #3 models
> show #3 models
> color #3 bychain
> show #2 models
> show #1 models
> hide #1 models
> hide #2 models
> hide #3 models
> show #2 models
> color #2 bypolymer
> color #2 bychain
> color #2 #00aa7fff
> color #2 #aa55ffff
> color #2 #aa0000ff
> color #2 #aa007fff
> color #2 #55aaffff
> color #2 bychain
> show #3 models
> hide #2 models
> matchmaker #3 to #2
Parameters
---
Chain pairing | bb
Alignment algorithm | Needleman-Wunsch
Similarity matrix | BLOSUM-62
SS fraction | 0.3
Gap open (HH/SS/other) | 18/18/6
Gap extend | 1
SS matrix | | | H | S | O
---|---|---|---
H | 6 | -9 | -6
S | | 6 | -6
O | | | 4
Iteration cutoff | 2
Matchmaker CAD_dimer_Deepmind_selected_prediction.pdb, chain A (#2) with
CAD_iCAD_dimer_Deepmind_selected_prediction.pdb, chain A (#3), sequence
alignment score = 1719.1
RMSD between 86 pruned atom pairs is 0.543 angstroms; (across all 338 pairs:
30.269)
> show #2 models
> hide #2 models
> show #2 models
> hide #2 models
> show #2 models
> hide #2 models
> show #2 models
> hide #2 models
> show #2 models
> hide #2 models
> hide #3 models
> show #2 models
> show #1 models
> hide #2 models
> show #1 surfaces
> show #2 models
> hide #2 models
> show #3 models
> matchmaker #3 to #1
Parameters
---
Chain pairing | bb
Alignment algorithm | Needleman-Wunsch
Similarity matrix | BLOSUM-62
SS fraction | 0.3
Gap open (HH/SS/other) | 18/18/6
Gap extend | 1
SS matrix | | | H | S | O
---|---|---|---
H | 6 | -9 | -6
S | | 6 | -6
O | | | 4
Iteration cutoff | 2
Matchmaker AF-O76075-F1-model_v2.pdb, chain A (#1) with
CAD_iCAD_dimer_Deepmind_selected_prediction.pdb, chain A (#3), sequence
alignment score = 1713.1
RMSD between 79 pruned atom pairs is 0.270 angstroms; (across all 338 pairs:
34.008)
> matchmaker #3 to #1:A
No 'to' model specified
> matchmaker #3 to #1
Parameters
---
Chain pairing | bb
Alignment algorithm | Needleman-Wunsch
Similarity matrix | BLOSUM-62
SS fraction | 0.3
Gap open (HH/SS/other) | 18/18/6
Gap extend | 1
SS matrix | | | H | S | O
---|---|---|---
H | 6 | -9 | -6
S | | 6 | -6
O | | | 4
Iteration cutoff | 2
Matchmaker AF-O76075-F1-model_v2.pdb, chain A (#1) with
CAD_iCAD_dimer_Deepmind_selected_prediction.pdb, chain A (#3), sequence
alignment score = 1713.1
RMSD between 79 pruned atom pairs is 0.270 angstroms; (across all 338 pairs:
34.008)
> select add #1
2751 atoms, 2811 bonds, 338 residues, 1 model selected
> hide sel surfaces
> select add #2
13731 atoms, 13911 bonds, 1014 residues, 3 models selected
> select subtract #1
10980 atoms, 11100 bonds, 676 residues, 2 models selected
> select add #3
21577 atoms, 21787 bonds, 1345 residues, 2 models selected
> select subtract #2
10597 atoms, 10687 bonds, 669 residues, 1 model selected
> select subtract #3
Nothing selected
> hide #3 models
> show #2 models
> select add #2
10980 atoms, 11100 bonds, 676 residues, 1 model selected
> select subtract #2
Nothing selected
> select add #1
2751 atoms, 2811 bonds, 338 residues, 1 model selected
> open C:/MW/MW_2022/Arnim/antibody_project/1r70(1).pdb
1r70(1).pdb title:
Model of human IGA2 determined by solution scattering, curve fitting and
homology modelling [more info...]
Chain information for 1r70(1).pdb #4
---
Chain | Description
A C | human IGA2(M1) light chain
B D | human IGA2(M1) heavy chain
> hide #!4 models
> show #!4 models
> hide #!4 models
> show #!4 models
> matchmaker #4 to #1
Parameters
---
Chain pairing | bb
Alignment algorithm | Needleman-Wunsch
Similarity matrix | BLOSUM-62
SS fraction | 0.3
Gap open (HH/SS/other) | 18/18/6
Gap extend | 1
SS matrix | | | H | S | O
---|---|---|---
H | 6 | -9 | -6
S | | 6 | -6
O | | | 4
Iteration cutoff | 2
Matchmaker AF-O76075-F1-model_v2.pdb, chain A (#1) with 1r70(1).pdb, chain B
(#4), sequence alignment score = 30
RMSD between 7 pruned atom pairs is 1.028 angstroms; (across all 29 pairs:
32.437)
> show sel surfaces
> select subtract #1
1 model selected
> show #3 models
> show surfaces
> hide #4.2 models
> hide #4.3 models
> hide #4.4 models
> hide #4.5 models
> color bychain
> hide #!4 models
> show #!4 models
> hide #!3 models
> show #!3 models
> hide #!2 models
> hide #!1 models
> show #!1 models
> hide #!3 models
> show #!3 models
> hide #!3 models
> show #!3 models
> hide #!3 models
> hide #!1 models
> hide #1.1 models
> color #1 #00aaffff
> show #!1 models
> show #1.1 models
> hide #1.1 models
> show #1.1 models
> hide #1.1 models
> show #!2 models
> hide #2.1 models
> hide #2.2 models
> show #!3 models
> hide #!3 models
> show #!3 models
> hide #!3 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!1 models
> show #!1 models
> hide #!2 models
> show #!2 models
> show #2.1 models
> hide #2.1 models
> show #2.2 models
> hide #2.2 models
> show #2.2 models
> hide #2.2 models
> save C:/MW/MW_2022/Arnim/antibody_project/CAD_CAD2_CADiCAD_Fab-full.cxs
> lighting full
> save C:/MW/MW_2022/Arnim/antibody_project/CAD_CAD2_CADiCAD_Fab-full.cxs
——— End of log from Fri Feb 10 17:20:21 2023 ———
opened ChimeraX session
> show #!3 models
> hide #3.1 models
> hide #3.2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> hide #!1 models
> show #!1 models
> hide #!1 models
> show #!1 models
> hide #!1 models
> show #!1 models
> movie record
> turn y 2 180
> wait 180
> movie encode C:\Users\weber\Desktop\movie1.mp4
Movie saved to \C:Users\\...\Desktop\movie1.mp4
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> rename #4 1r70.pdb
> lighting full
> show surfaces
> hide surfaces
> select add #4.2
214 atoms, 214 residues, 1 model selected
> select subtract #4.2
1 model selected
> select add #4.2
214 atoms, 214 residues, 1 model selected
> select subtract #4.2
1 model selected
> select add #4.2
214 atoms, 214 residues, 1 model selected
> hide #!2 models
> show #!2 models
> hide #!3 models
> hide #!4 models
> show #!4 models
> show #!3 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> select #3/B
5107 atoms, 5137 bonds, 331 residues, 1 model selected
> hide #!4 models
> hide #!2 models
> show #!2 models
> hide #2.1 models
> show #2.1 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> matchmaker #3 to #1
Parameters
---
Chain pairing | bb
Alignment algorithm | Needleman-Wunsch
Similarity matrix | BLOSUM-62
SS fraction | 0.3
Gap open (HH/SS/other) | 18/18/6
Gap extend | 1
SS matrix | | | H | S | O
---|---|---|---
H | 6 | -9 | -6
S | | 6 | -6
O | | | 4
Iteration cutoff | 2
Matchmaker AF-O76075-F1-model_v2.pdb, chain A (#1) with
CAD_iCAD_dimer_Deepmind_selected_prediction.pdb, chain A (#3), sequence
alignment score = 1713.1
RMSD between 79 pruned atom pairs is 0.270 angstroms; (across all 338 pairs:
34.008)
> matchmaker #3 to #2
Parameters
---
Chain pairing | bb
Alignment algorithm | Needleman-Wunsch
Similarity matrix | BLOSUM-62
SS fraction | 0.3
Gap open (HH/SS/other) | 18/18/6
Gap extend | 1
SS matrix | | | H | S | O
---|---|---|---
H | 6 | -9 | -6
S | | 6 | -6
O | | | 4
Iteration cutoff | 2
Matchmaker CAD_dimer_Deepmind_selected_prediction.pdb, chain A (#2) with
CAD_iCAD_dimer_Deepmind_selected_prediction.pdb, chain A (#3), sequence
alignment score = 1719.1
RMSD between 86 pruned atom pairs is 0.543 angstroms; (across all 338 pairs:
30.269)
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> show #!2 models
> ui tool show "Show Sequence Viewer"
> sequence chain #3/B
Alignment identifier is 3/B
Traceback (most recent call last):
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\show_sequences\tool.py", line 64, in show_seqs
run(self.session, "seq chain %s" % " ".join([chain.atomspec for chain in
chains]))
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\core\commands\run.py", line 38, in run
results = command.run(text, log=log, return_json=return_json)
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\core\commands\cli.py", line 2897, in run
result = ci.function(session, **kw_args)
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\seqalign\cmd.py", line 212, in seqalign_chain
alignment = session.alignments.new_alignment([chain], ident, seq_viewer="sv",
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\seqalign\manager.py", line 242, in new_alignment
alignment = Alignment(self.session, seqs, identify_as, attrs, markups,
auto_destroy,
TypeError: __init__() got an unexpected keyword argument 'seq_viewer'
TypeError: __init__() got an unexpected keyword argument 'seq_viewer'
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\seqalign\manager.py", line 242, in new_alignment
alignment = Alignment(self.session, seqs, identify_as, attrs, markups,
auto_destroy,
See log for complete Python traceback.
> ui tool show "Show Sequence Viewer"
> sequence chain #1/A #2/A #2/B #3/A
Alignment identifier is 1
Traceback (most recent call last):
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\show_sequences\tool.py", line 64, in show_seqs
run(self.session, "seq chain %s" % " ".join([chain.atomspec for chain in
chains]))
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\core\commands\run.py", line 38, in run
results = command.run(text, log=log, return_json=return_json)
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\core\commands\cli.py", line 2897, in run
result = ci.function(session, **kw_args)
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\seqalign\cmd.py", line 232, in seqalign_chain
alignment = session.alignments.new_alignment([seq], None, seq_viewer="sv",
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\seqalign\manager.py", line 242, in new_alignment
alignment = Alignment(self.session, seqs, identify_as, attrs, markups,
auto_destroy,
TypeError: __init__() got an unexpected keyword argument 'seq_viewer'
TypeError: __init__() got an unexpected keyword argument 'seq_viewer'
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\seqalign\manager.py", line 242, in new_alignment
alignment = Alignment(self.session, seqs, identify_as, attrs, markups,
auto_destroy,
See log for complete Python traceback.
> ui tool show "Show Sequence Viewer"
> sequence chain #1/A
Alignment identifier is 1/A
Traceback (most recent call last):
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\show_sequences\tool.py", line 64, in show_seqs
run(self.session, "seq chain %s" % " ".join([chain.atomspec for chain in
chains]))
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\core\commands\run.py", line 38, in run
results = command.run(text, log=log, return_json=return_json)
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\core\commands\cli.py", line 2897, in run
result = ci.function(session, **kw_args)
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\seqalign\cmd.py", line 212, in seqalign_chain
alignment = session.alignments.new_alignment([chain], ident, seq_viewer="sv",
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\seqalign\manager.py", line 242, in new_alignment
alignment = Alignment(self.session, seqs, identify_as, attrs, markups,
auto_destroy,
TypeError: __init__() got an unexpected keyword argument 'seq_viewer'
TypeError: __init__() got an unexpected keyword argument 'seq_viewer'
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\seqalign\manager.py", line 242, in new_alignment
alignment = Alignment(self.session, seqs, identify_as, attrs, markups,
auto_destroy,
See log for complete Python traceback.
> hide #1.1 models
> hide #2.1 models
> hide #2.2 models
> hide #3.1 models
> hide #3.2 models
> ui tool show "Show Sequence Viewer"
> sequence chain #1/A #2/A #2/B #3/A
Alignment identifier is 1
Traceback (most recent call last):
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\show_sequences\tool.py", line 64, in show_seqs
run(self.session, "seq chain %s" % " ".join([chain.atomspec for chain in
chains]))
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\core\commands\run.py", line 38, in run
results = command.run(text, log=log, return_json=return_json)
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\core\commands\cli.py", line 2897, in run
result = ci.function(session, **kw_args)
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\seqalign\cmd.py", line 232, in seqalign_chain
alignment = session.alignments.new_alignment([seq], None, seq_viewer="sv",
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\seqalign\manager.py", line 242, in new_alignment
alignment = Alignment(self.session, seqs, identify_as, attrs, markups,
auto_destroy,
TypeError: __init__() got an unexpected keyword argument 'seq_viewer'
TypeError: __init__() got an unexpected keyword argument 'seq_viewer'
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\seqalign\manager.py", line 242, in new_alignment
alignment = Alignment(self.session, seqs, identify_as, attrs, markups,
auto_destroy,
See log for complete Python traceback.
> ui tool show "Show Sequence Viewer"
> sequence chain #1/A
Alignment identifier is 1/A
Traceback (most recent call last):
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\show_sequences\tool.py", line 64, in show_seqs
run(self.session, "seq chain %s" % " ".join([chain.atomspec for chain in
chains]))
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\core\commands\run.py", line 38, in run
results = command.run(text, log=log, return_json=return_json)
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\core\commands\cli.py", line 2897, in run
result = ci.function(session, **kw_args)
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\seqalign\cmd.py", line 212, in seqalign_chain
alignment = session.alignments.new_alignment([chain], ident, seq_viewer="sv",
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\seqalign\manager.py", line 242, in new_alignment
alignment = Alignment(self.session, seqs, identify_as, attrs, markups,
auto_destroy,
TypeError: __init__() got an unexpected keyword argument 'seq_viewer'
TypeError: __init__() got an unexpected keyword argument 'seq_viewer'
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\seqalign\manager.py", line 242, in new_alignment
alignment = Alignment(self.session, seqs, identify_as, attrs, markups,
auto_destroy,
See log for complete Python traceback.
OpenGL version: 3.3.14831 Core Profile Forward-Compatible Context 21.5.2 27.20.20903.8001
OpenGL renderer: AMD Radeon R9 200 Series
OpenGL vendor: ATI Technologies Inc.
Python: 3.9.11
Locale: de_DE.cp1252
Qt version: PyQt6 6.4.2, Qt 6.4.2
Qt runtime version: 6.4.2
Qt platform: windows
Manufacturer: Gigabyte Technology Co., Ltd.
Model: H67MA-UD2H
OS: Microsoft Windows 10 Pro (Build 19044)
Memory: 17,162,481,664
MaxProcessMemory: 137,438,953,344
CPU: 4 Intel(R) Core(TM) i5-2500K CPU @ 3.30GHz
OSLanguage: de-DE
Installed Packages:
alabaster: 0.7.13
appdirs: 1.4.4
asttokens: 2.2.1
Babel: 2.11.0
backcall: 0.2.0
beautifulsoup4: 4.11.2
blockdiag: 3.0.0
build: 0.10.0
certifi: 2022.12.7
cftime: 1.6.2
charset-normalizer: 3.0.1
ChimeraX-AddCharge: 1.5.8
ChimeraX-AddH: 2.2.3
ChimeraX-AlignmentAlgorithms: 2.0.1
ChimeraX-AlignmentHdrs: 3.3.1
ChimeraX-AlignmentMatrices: 2.1
ChimeraX-Alignments: 2.9
ChimeraX-AlphaFold: 1.0
ChimeraX-AltlocExplorer: 1.0.3
ChimeraX-AmberInfo: 1.0
ChimeraX-Arrays: 1.1
ChimeraX-Atomic: 1.43.7
ChimeraX-AtomicLibrary: 10.0.3
ChimeraX-AtomSearch: 2.0.1
ChimeraX-AxesPlanes: 2.3.2
ChimeraX-BasicActions: 1.1.2
ChimeraX-BILD: 1.0
ChimeraX-BlastProtein: 2.1.2
ChimeraX-BondRot: 2.0.1
ChimeraX-BugReporter: 1.0.1
ChimeraX-BuildStructure: 2.8
ChimeraX-Bumps: 1.0
ChimeraX-BundleBuilder: 1.2.1
ChimeraX-ButtonPanel: 1.0.1
ChimeraX-CageBuilder: 1.0.1
ChimeraX-CellPack: 1.0
ChimeraX-Centroids: 1.3.2
ChimeraX-ChangeChains: 1.0.2
ChimeraX-CheckWaters: 1.3.1
ChimeraX-ChemGroup: 2.0.1
ChimeraX-Clashes: 2.2.4
ChimeraX-ColorActions: 1.0.3
ChimeraX-ColorGlobe: 1.0
ChimeraX-ColorKey: 1.5.3
ChimeraX-CommandLine: 1.2.5
ChimeraX-ConnectStructure: 2.0.1
ChimeraX-Contacts: 1.0.1
ChimeraX-Core: 1.6.dev202302100235
ChimeraX-CoreFormats: 1.1
ChimeraX-coulombic: 1.4.2
ChimeraX-Crosslinks: 1.0
ChimeraX-Crystal: 1.0
ChimeraX-CrystalContacts: 1.0.1
ChimeraX-DataFormats: 1.2.3
ChimeraX-Dicom: 1.1
ChimeraX-DistMonitor: 1.3.3
ChimeraX-DockPrep: 1.1
ChimeraX-Dssp: 2.0
ChimeraX-EMDB-SFF: 1.0
ChimeraX-ESMFold: 1.0
ChimeraX-ExperimentalCommands: 1.0
ChimeraX-FileHistory: 1.0.1
ChimeraX-FunctionKey: 1.0.1
ChimeraX-Geometry: 1.3
ChimeraX-gltf: 1.0
ChimeraX-Graphics: 1.1.1
ChimeraX-Hbonds: 2.4
ChimeraX-Help: 1.2.1
ChimeraX-HKCage: 1.3
ChimeraX-IHM: 1.1
ChimeraX-ImageFormats: 1.2
ChimeraX-IMOD: 1.0
ChimeraX-IO: 1.0.1
ChimeraX-ItemsInspection: 1.0.1
ChimeraX-Label: 1.1.7
ChimeraX-ListInfo: 1.1.1
ChimeraX-Log: 1.1.5
ChimeraX-LookingGlass: 1.1
ChimeraX-Maestro: 1.8.2
ChimeraX-Map: 1.1.4
ChimeraX-MapData: 2.0
ChimeraX-MapEraser: 1.0.1
ChimeraX-MapFilter: 2.0.1
ChimeraX-MapFit: 2.0
ChimeraX-MapSeries: 2.1.1
ChimeraX-Markers: 1.0.1
ChimeraX-Mask: 1.0.2
ChimeraX-MatchMaker: 2.0.11
ChimeraX-MDcrds: 2.6
ChimeraX-MedicalToolbar: 1.0.2
ChimeraX-Meeting: 1.0.1
ChimeraX-MLP: 1.1.1
ChimeraX-mmCIF: 2.11
ChimeraX-MMTF: 2.2
ChimeraX-Modeller: 1.5.8
ChimeraX-ModelPanel: 1.3.6
ChimeraX-ModelSeries: 1.0.1
ChimeraX-Mol2: 2.0
ChimeraX-Mole: 1.0
ChimeraX-Morph: 1.0.2
ChimeraX-MouseModes: 1.2
ChimeraX-Movie: 1.0
ChimeraX-Neuron: 1.0
ChimeraX-Nucleotides: 2.0.3
ChimeraX-OpenCommand: 1.10.1
ChimeraX-PDB: 2.6.13
ChimeraX-PDBBio: 1.0
ChimeraX-PDBLibrary: 1.0.2
ChimeraX-PDBMatrices: 1.0
ChimeraX-PickBlobs: 1.0.1
ChimeraX-Positions: 1.0
ChimeraX-PresetMgr: 1.1
ChimeraX-PubChem: 2.1
ChimeraX-ReadPbonds: 1.0.1
ChimeraX-Registration: 1.1.1
ChimeraX-RemoteControl: 1.0
ChimeraX-RenderByAttr: 1.0
ChimeraX-RenumberResidues: 1.1
ChimeraX-ResidueFit: 1.0.1
ChimeraX-RestServer: 1.1
ChimeraX-RNALayout: 1.0
ChimeraX-RotamerLibMgr: 3.0
ChimeraX-RotamerLibsDunbrack: 2.0
ChimeraX-RotamerLibsDynameomics: 2.0
ChimeraX-RotamerLibsRichardson: 2.0
ChimeraX-SaveCommand: 1.5.1
ChimeraX-SchemeMgr: 1.0
ChimeraX-SDF: 2.0.1
ChimeraX-Segger: 1.0
ChimeraX-Segment: 1.0.1
ChimeraX-SelInspector: 1.0
ChimeraX-SeqView: 2.8.1
ChimeraX-Shape: 1.0.1
ChimeraX-Shell: 1.0.1
ChimeraX-Shortcuts: 1.1.1
ChimeraX-ShowSequences: 1.0.1
ChimeraX-SideView: 1.0.1
ChimeraX-Smiles: 2.1
ChimeraX-SmoothLines: 1.0
ChimeraX-SpaceNavigator: 1.0
ChimeraX-StdCommands: 1.10.1
ChimeraX-STL: 1.0.1
ChimeraX-Storm: 1.0
ChimeraX-StructMeasure: 1.1.1
ChimeraX-Struts: 1.0.1
ChimeraX-Surface: 1.0.1
ChimeraX-SwapAA: 2.0.1
ChimeraX-SwapRes: 2.2.1
ChimeraX-TapeMeasure: 1.0
ChimeraX-Test: 1.0
ChimeraX-Toolbar: 1.1.2
ChimeraX-ToolshedUtils: 1.2.1
ChimeraX-Topography: 1.0
ChimeraX-Tug: 1.0.1
ChimeraX-UI: 1.27
ChimeraX-uniprot: 2.2.2
ChimeraX-UnitCell: 1.0.1
ChimeraX-ViewDockX: 1.2
ChimeraX-VIPERdb: 1.0
ChimeraX-Vive: 1.1
ChimeraX-VolumeMenu: 1.0.1
ChimeraX-VTK: 1.0
ChimeraX-WavefrontOBJ: 1.0
ChimeraX-WebCam: 1.0.2
ChimeraX-WebServices: 1.1.1
ChimeraX-Zone: 1.0.1
colorama: 0.4.6
comm: 0.1.2
comtypes: 1.1.14
contourpy: 1.0.7
cxservices: 1.2.2
cycler: 0.11.0
Cython: 0.29.33
debugpy: 1.6.6
decorator: 5.1.1
docutils: 0.19
executing: 1.2.0
filelock: 3.9.0
fonttools: 4.38.0
funcparserlib: 1.0.1
grako: 3.16.5
h5py: 3.8.0
html2text: 2020.1.16
idna: 3.4
ihm: 0.35
imagecodecs: 2022.9.26
imagesize: 1.4.1
importlib-metadata: 6.0.0
ipykernel: 6.21.1
ipython: 8.9.0
ipython-genutils: 0.2.0
ipywidgets: 8.0.4
jedi: 0.18.2
Jinja2: 3.1.2
jupyter-client: 8.0.2
jupyter-core: 5.2.0
jupyterlab-widgets: 3.0.5
kiwisolver: 1.4.4
line-profiler: 4.0.2
lxml: 4.9.2
lz4: 4.3.2
MarkupSafe: 2.1.2
matplotlib: 3.6.3
matplotlib-inline: 0.1.6
msgpack: 1.0.4
nest-asyncio: 1.5.6
netCDF4: 1.6.2
networkx: 2.8.8
numexpr: 2.8.4
numpy: 1.23.5
openvr: 1.23.701
packaging: 23.0
pandas: 1.5.3
ParmEd: 3.4.3
parso: 0.8.3
pep517: 0.13.0
pickleshare: 0.7.5
Pillow: 9.3.0
pip: 23.0
pkginfo: 1.9.6
platformdirs: 3.0.0
prompt-toolkit: 3.0.36
psutil: 5.9.4
pure-eval: 0.2.2
pycollada: 0.7.2
pydicom: 2.3.0
Pygments: 2.14.0
PyOpenGL: 3.1.5
PyOpenGL-accelerate: 3.1.5
pyparsing: 3.0.9
pyproject-hooks: 1.0.0
PyQt6-commercial: 6.4.2
PyQt6-Qt6: 6.4.2
PyQt6-sip: 13.4.1
PyQt6-WebEngine-commercial: 6.4.0
PyQt6-WebEngine-Qt6: 6.4.2
python-dateutil: 2.8.2
pytz: 2022.7.1
pywin32: 305
pyzmq: 25.0.0
qtconsole: 5.4.0
QtPy: 2.3.0
RandomWords: 0.4.0
requests: 2.28.2
scipy: 1.9.3
setuptools: 65.1.1
sfftk-rw: 0.7.3
six: 1.16.0
snowballstemmer: 2.2.0
sortedcontainers: 2.4.0
soupsieve: 2.3.2.post1
sphinx: 6.1.3
sphinx-autodoc-typehints: 1.22
sphinxcontrib-applehelp: 1.0.4
sphinxcontrib-blockdiag: 3.0.0
sphinxcontrib-devhelp: 1.0.2
sphinxcontrib-htmlhelp: 2.0.1
sphinxcontrib-jsmath: 1.0.1
sphinxcontrib-qthelp: 1.0.3
sphinxcontrib-serializinghtml: 1.1.5
stack-data: 0.6.2
tables: 3.7.0
tcia-utils: 1.0.2
tifffile: 2022.10.10
tinyarray: 1.2.4
tomli: 2.0.1
tornado: 6.2
traitlets: 5.9.0
urllib3: 1.26.14
wcwidth: 0.2.6
webcolors: 1.12
wheel: 0.38.4
wheel-filename: 1.4.1
widgetsnbextension: 4.0.5
WMI: 1.5.1
zipp: 3.13.0
Attachments (2)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Change History (13)
comment:1 by , 3 years ago
| Component: | Unassigned → Sequence |
|---|---|
| Milestone: | → 1.6 |
| Owner: | set to |
| Platform: | → all |
| Priority: | normal → major |
| Project: | → ChimeraX |
| Status: | new → accepted |
| Summary: | ChimeraX bug report submission → Show chain sequence: TypeError: __init__() got an unexpected keyword argument 'seq_viewer' |
comment:2 by , 3 years ago
comment:3 by , 3 years ago
| Resolution: | → fixed |
|---|---|
| Status: | accepted → closed |
Hi Michael,
Thanks for reporting the problem with the sequence viewer. I have fixed it and the fix will be in the next daily build. As for the SwissDock / model panel problem, how are you using SwissDock in 1.5? Did you somehow transplant the revised viewdockx module into your 1.5 installation?
--Eric
Eric Pettersen
UCSF Computer Graphics Lab
follow-up: 4 comment:4 by , 3 years ago
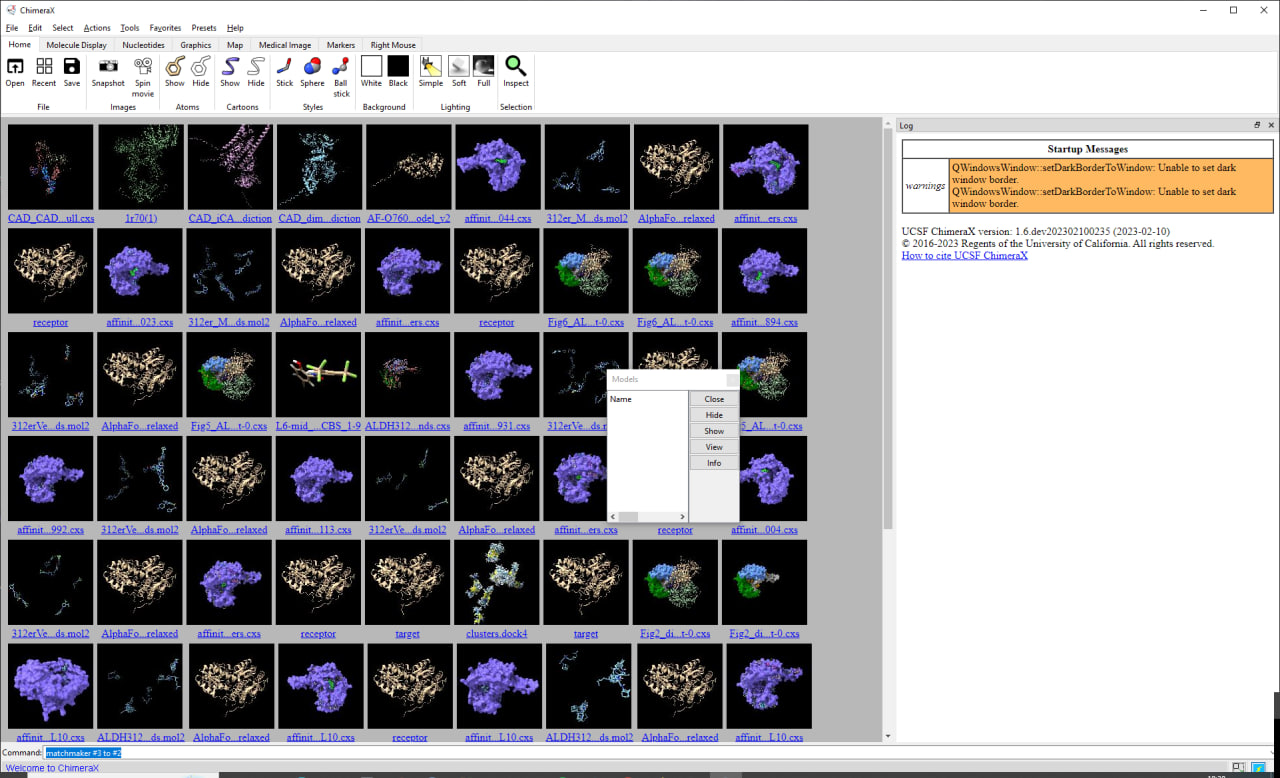
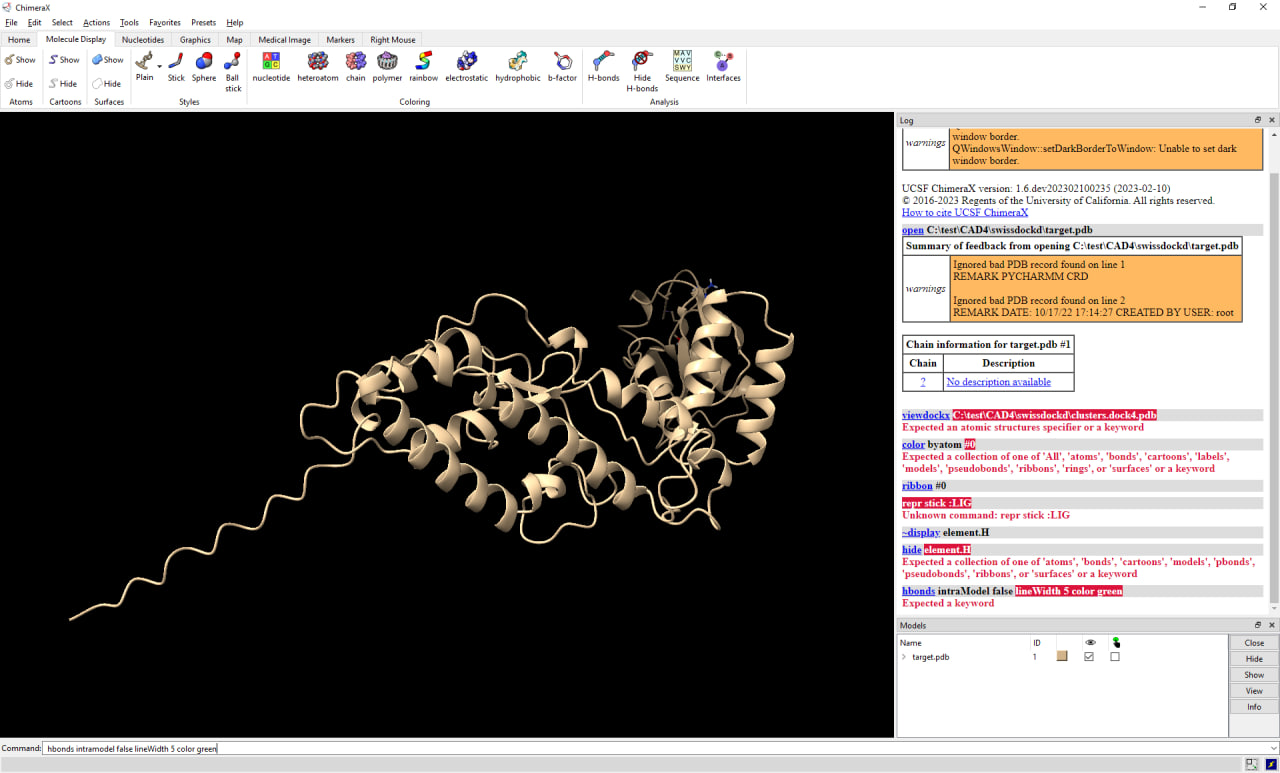
Hi Eric, I had reported the non-functional viewdockx module in ChimeraX (when used with SwissDock output files) about 2 years ago (see bug report #4232). Following the ChimeraX update notifications (see ChimeraX notification email from 2023/02/03 on bug #4232) I had actually assumed that viewdockx had been repaired and was functional. When trying with ChimeraX 1.5 I noticed that it still does not work plus that the daily build I used for testing (2023/02/09) caused an additional bug with permanent alteration of the setup of the "Tools/Models" panel as reported a few days ago (see bug report #8473). For the latter, please take a look at the attached screenshot (photo_2023-02-10_18-39-07_ChimeraX-Startup.jpg). Regarding the former, please see the second screenshot (photo_2023-02-14_16-56-37_viewdockx.jpg). The script I used to try loading the SwissDock docking output files is the following one which I got from Elaine Meng a long time ago when I first asked about how to work with SwissDock in ChimeraX (and not Chimera): open C:\test\CAD4\swissdockd\target.pdb viewdockx C:\test\CAD4\swissdockd\clusters.dock4.pdb color byatom #0 ribbon #0 repr stick :LIG ~disp element.H hbonds intramodel false lineWidth 5 color green Note that all the files I used as well as this script work fine with Chimera 1.16. Also, manual loading of the target protein plus the clusters.dock4.pdb file containing the aggregated ligands works fine in ChimeraX. Cycling through the individual ligands plus displaying the interaction properties as possible in Chimera 1.1.6 however so far remains nonfunctional. Best regards, Michael. Am 14.02.2023 02:52 schrieb ChimeraX:
by , 3 years ago
| Attachment: | photo_2023-02-10_18-39-07_ChimeraX-Startup.jpg added |
|---|
Added by email2trac
comment:5 by , 3 years ago
Hi Michael,
As per the notification on bug #4232, SwissDock support is only in the daily build, not 1.5. Once released, a production release is never changed. If changes are needed, there would be a different version number (e.g. 1.5.1 or 1.6).
As for your problem with the Model Panel, can you dock the Model Panel into the main window after startup? If you can, use the Model Panel's context menu (right click over the Model Panel) and choose "Save Tool Position". If you can't dock it, use its context menu and make sure "Dockable tool" is checked. Then save the model panel position.
As for the script commands, none of them are right for ChimeraX. The first two should be:
open C:\test\CAD4\swissdockd\target.pdb C:\test\CAD4\swissdockd\clusters.dock4.pdb format swissdock
viewdockx #2
The equivalent commands for the next five are "color", "cartoon", "stye", "hide", and "hbonds" respectively, but with different arguments. You can use "help <comamnd name>" to see the help page for a command and see what arguments it accepts.
--Eric
comment:6 by , 3 years ago
Hi Eric, actually, nothing of all that works. ;-) 1. I can dock the model panel into the main window (the right panel?) after it initially appeared as separate window upon startup. What I can't do is saving this situation in Windows 10 - no idea where I should find that right-clickable saving option... 2. The commands you gave don't work: The first is ok, it loads the protein, the second just gives an error message: "No suitable models found for ViewDockX" -Michael. Am 14.02.2023 20:18 schrieb ChimeraX:
comment:7 by , 3 years ago
Hi Michael,
1) Once you dock the Model Panel, you need to right click over it -- that will bring up a context menu with "Save Tool Position" in it.
2) Once you use that open command, is clusters.dock4.pdb model #2? If not, use it's model number in the viewdockx command rather than "#2".
--Eric
Eric Pettersen
UCSF Computer Graphics Lab
follow-up: 6 comment:8 by , 3 years ago
Hi Eric, (1) I tried the "Save Tool Position" - it simply won't work. (2) Regarding the viewdockx it finally works (forgot the open command when loading the second pdb file (ligands)). Thanks a lot, Michael. Am 22.02.2023 18:04 schrieb ChimeraX:
comment:9 by , 3 years ago
Hi Michael,
By "simply won't work" do you mean you can't get the context menu, or that using "Save Tool Position" doesn't fix the problem?
--Eric
follow-up: 8 comment:10 by , 3 years ago
...the latter. I found that option, I clicked on it, unfortunately - it has no effect. -Michael. Am 24.02.2023 20:41 schrieb ChimeraX:
comment:11 by , 3 years ago
Does saving _any_ tool positions work? For instance, drag the log out of the main window so it's floating. Choose "Save Tool Position" on it. Stop and restart. Does the log now start out floating or does it start out docked?
--Eric
Reported by Michael Weber