Opened 9 years ago
Closed 8 years ago
#642 closed defect (fixed)
OpenMM bug
| Reported by: | Owned by: | ||
|---|---|---|---|
| Priority: | normal | Milestone: | |
| Component: | Structure Editing | Version: | |
| Keywords: | Cc: | chimera-programmers@… | |
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
Hi all, Please take a look at the issue I've just posted on the OpenMM Github site: https://github.com/pandegroup/openmm/issues/1790. Something's all of a sudden going badly wrong with how OpenMM behaves when I have map forces included. Granted I haven't used it much over the last few months while I was working on the Clipper plugin, but I haven't seen any behaviour like it before. Not sure if I just got lucky with the test cases I chose previously, or if the move to Python 3.6 has subtly broken things. I do see there's an updated OpenMM version out (7.1 - we're on 7.0.1) - it might be worth upgrading in case that fixes it? Thanks, Tristan Tristan Croll Research Fellow Cambridge Institute for Medical Research University of Cambridge CB2 0XY
Attachments (3)
{kind=link}
{kind=link}
Change History (29)
comment:1 by , 9 years ago
| Cc: | added |
|---|---|
| Component: | Unassigned → Structure Editing |
| Owner: | set to |
| Platform: | → all |
| Project: | → ChimeraX |
| Status: | new → assigned |
comment:2 by , 9 years ago
follow-up: 3 comment:3 by , 9 years ago
I'm afraid it shows up on another (previously working) model as well. I
just have no idea if I was simply lucky enough to avoid the issue
previously, or if something has fundamentally changed.
Anyway, after a surprising amount of wailing and gnashing of teeth, I
have OpenMM 7.1.0 installed in ChimeraX. Not really the fault of OpenMM
- it turns out distutils in Python 3.6 is a bit broken (for my Fedora
system, at least). In brief, in distutils/unixccompiler.py:
def runtime_library_dir_option(self, dir):
# XXX Hackish, at the very least. See Python bug #445902:
# http://sourceforge.net/tracker/index.php
# ?func=detail&aid=445902&group_id=5470&atid=105470
# Linkers on different platforms need different options to
# specify that directories need to be added to the list of
# directories searched for dependencies when a dynamic library
# is sought. GCC on GNU systems (Linux, FreeBSD, ...) has to
# be told to pass the -R option through to the linker, whereas
# other compilers and gcc on other systems just know this.
# Other compilers may need something slightly different. At
# this time, there's no way to determine this information from
# the configuration data stored in the Python installation, so
# we use this hack.
compiler = os.path.basename(sysconfig.get_config_var("CC"))
if sys.platform[:6] == "darwin":
# MacOSX's linker doesn't understand the -R flag at all
return "-L" + dir
elif sys.platform[:7] == "freebsd":
return "-Wl,-rpath=" + dir
elif sys.platform[:5] == "hp-ux":
if self._is_gcc(compiler):
return ["-Wl,+s", "-L" + dir]
return ["+s", "-L" + dir]
elif sys.platform[:7] == "irix646" or sys.platform[:6] ==
"osf1V5":
return ["-rpath", dir]
else:
if self._is_gcc(compiler):
# gcc on non-GNU systems does not need -Wl, but can
# use it anyway. Since distutils has always passed in
# -Wl whenever gcc was used in the past it is probably
# safest to keep doing so.
if sysconfig.get_config_var("GNULD") == "yes":
# GNU ld needs an extra option to get a RUNPATH
# instead of just an RPATH.
return "-Wl,--enable-new-dtags,-R" + dir
else:
return "-Wl,-R" + dir
else:
# No idea how --enable-new-dtags would be passed on to
# ld if this system was using GNU ld. Don't know if a
# system like this even exists.
return "-R" + dir
Suffice to say, on my machine it finds its way to the "No idea how..."
option, because sysconfig.get_config_var("CC") returns something it
clearly wasn't expecting:
'gcc -pipe -fPIC -std=gnu99 -I/tmp/chx-develop.BXqiYX/build/include
-L/tmp/chx-develop.BXqiYX/build/lib -DUSE_DYLD_GLOBAL_NAMESPACE
-pthread'
... which os.path.basename helpfully strips back to:
'lib -DUSE_DYLD_GLOBAL_NAMESPACE -pthread'
... rather than the 'gcc' it's looking for. My very hackish workaround
was to just replace "-R" (which crashes the C++ compiler) with "-Wl,-R"
in the last line.
Will see if it's actually fixed anything now.
On 2017-04-21 23:59, ChimeraX wrote:
follow-up: 4 comment:4 by , 9 years ago
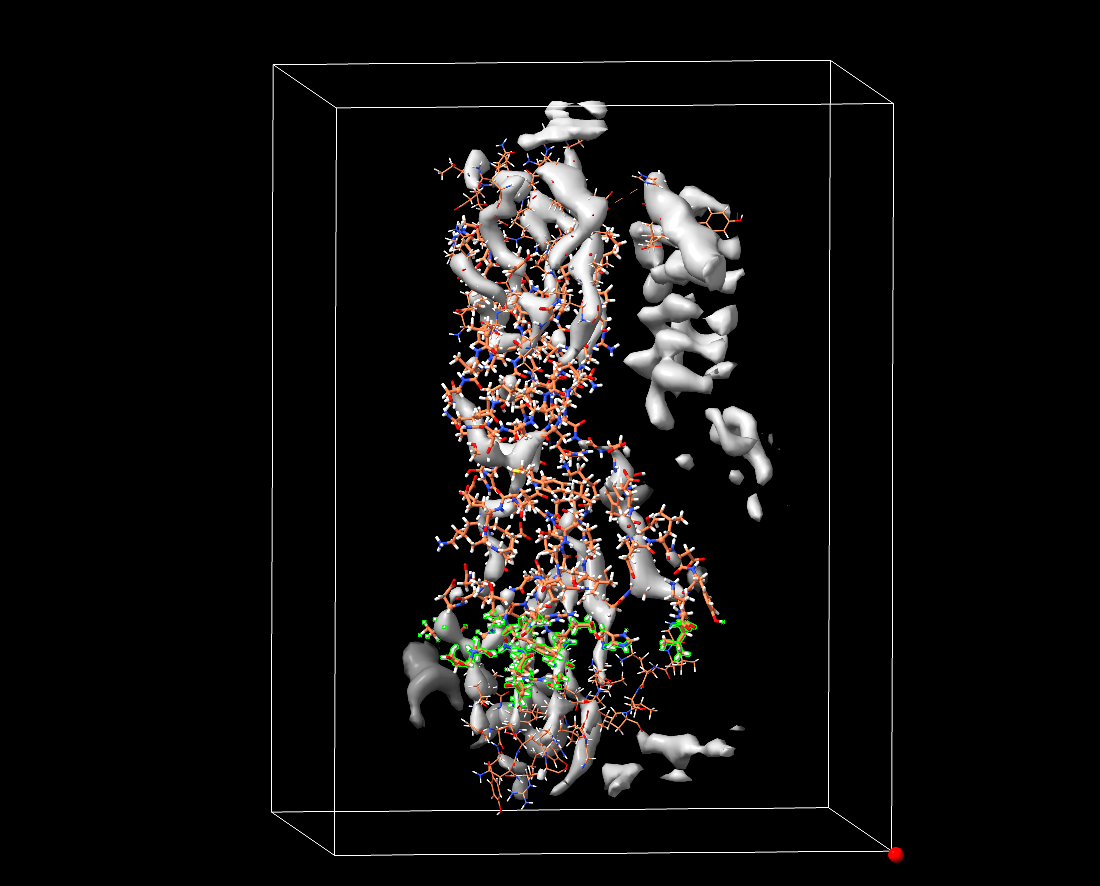
The update to 7.1.0 didn't fix anything. It's quite clearly a race condition within the OpenMM core. See attached: the box shows the extent of the volume used to generate the Continuous3DForce, with the red sphere at maximum (i,j,k). The thicker atoms are the mobile ones, with the thicker ones fixed (so excluded from map forces). The selected atoms are all experiencing forces of +/- 2.14748 kJ/mol/nm on each axis (i.e. they've been fed uninitialised values). I've posted as such to the OpenMM Github - looks like it will be up to them to fix it. On 2017-04-21 23:59, ChimeraX wrote:
comment:5 by , 9 years ago
Update: I've boiled it down to an isolated test case (see https://github.com/pandegroup/openmm/issues/1790#issuecomment-298594895).
comment:6 by , 9 years ago
Solved, I think. The good news is it's not a fundamental bug in OpenMM itself, just in their SWIG wrapping. The Continuous3DFunction takes the array input as const vector<double>&, and somebody messed up the typemapping for this (and a lot of other const reference typemaps as well, by the look of it). Basically, their typemap creates a vector from the Python input array and returns a reference to it, but the vector goes out of scope the moment the constructor completes. Not good.
comment:7 by , 9 years ago
Belay that - my misunderstanding. It makes its own copy of the vector, which stays safely in scope.
comment:8 by , 9 years ago
Found the issue (for real this time). Somebody's bad copy-paste in the 3D spline generation code (SplineFitter.cpp):
double deltax = x[nexti]-x[i];
double deltay = y[nextj]-y[j];
< double deltaz = z[nextj]-z[j];
---
> double deltaz = z[nextk]-z[k];
comment:9 by , 9 years ago
I've re-compiled OpenMM and confirmed that this was the problem - which leaves me with a big favour to ask. I'm scheduled to give a demonstration of ISOLDE at the Erice School of Crystallography in the first week of June, and that's going to be a little problematic if this bug is still in the OpenMM build incorporated into ChimeraX. If I absolutely have to I can work around it for the time being by ensuring I only ever pass OpenMM volumes with equal dimensions and spacing on the y and z axes, but that would obviously be quite the kludge. Is there any chance of getting a patched build of OpenMM into the official ChimeraX distribution by that time?
comment:10 by , 9 years ago
Hi Tristan, We have never compiled OpenMM, just included the binary distributions in ChimeraX. I recall it looked like a real pain to build. Do you need the fixed version on Windows, Mac and Linux? Does it need CUDA and OpenCL support? Here are some Windows OpenMM build instructions. https://github.com/pandegroup/openmm/wiki/Compile-OpenMM-7.0.1-on-Windows-8.1 <https://github.com/pandegroup/openmm/wiki/Compile-OpenMM-7.0.1-on-Windows-8.1> Can you make the needed builds and I will put them into ChimeraX as binary distributions? Or is the idea you want us to build this monster? This is a wake-up call, reminding us that OpenMM is not actively developed or actively maintained — and we will need to think carefully about the future. We can’t become the maintainers of a complex package like OpenMM, no matter how useful and irreplacable it is. But we’ll figure out something for your workshop. Tom
follow-up: 10 comment:11 by , 9 years ago
OK, I made builds for Mac and Linux at https://drive.google.com/open?id=0B6uMjfjuw4k8R2txLVBvZ29iREU and https://drive.google.com/open?id=0B6uMjfjuw4k8ZVlleFVYajU1Mnc respectively. It builds quite straightforwardly in these environments - the only real issue I found is that the latest version of Doxygen (8.0.13) segfaults during building on the Mac. Seems that's something new - they recommend 8.0.12 in their documentation, but I downgraded to 8.0.5 (which worked fine). It went essentially as described in the instructions at http://docs.openmm.org/7.1.0/userguide/library.html#compiling-openmm-from-source-code - make a build directory, ccmake, point it to the ChimeraX python executable, generate, and make.
After all that, though, I found that there are fresh builds for Linux and Mac available at https://anaconda.org/omnia/openmm/files. The 7.1.1 release candidates were just compiled today, and I've confirmed they have the bug fixed. They're built against Python 3.5, but from the behaviour of the existing version that shouldn't be a problem. If you choose to use these builds, though, could you replace site-packages/simtk/openmm/app/forcefield.py with the one in the existing ChimeraX distribution? It has the changes I need to allow simulations to run when residues are missing bonds.
It does seem like Windows is a very different beast, and I'm OK with avoiding it for the time being. The last Windows build on anaconda.org is about 3 months old. I did get a build to compile on Windows, but I don't trust it - the compile log is full of unsafe type cast warnings:
warning C4312: 'type cast': conversion from 'int' to 'void *' of greater size
Oh - and if you want the CUDA platform to work, you just need to have /usr/local/cuda/lib64 in the LD_LIBRARY_PATH (or DYLD_LIBRARY_PATH for Mac). It looks to be worth it - on my machine it's about 2-fold faster than the OpenCL platform.
comment:12 by , 9 years ago
Ok, I updated Linux and Mac OpenMM to 7.1.1 python 3.6 builds from https://anaconda.org/omnia/openmm/files <https://anaconda.org/omnia/openmm/files> and tested on Linux and Mac and both worked using my tug atoms mouse mode (seemed faster too). Will be in tonight’s ChimeraX build (built on May 10). There is no Windows OpenMM 7.1.1 build so that is still at 7.0.1. If participants at your June workshop are using Windows (usually a substantial fraction of people run Windows on their laptops), then we will need to figure out how to get a fixed OpenMM build for Windows. I eliminated the forcefield.py patch on Mac/Linux since it appears that the 7.1.1 forcefield.py has the changes from that patch, but you should verify this since the changes are a good bit different, for instance ignoreMissingExternalBonds from your patch seems to have become ignoreExternalBonds and many other differences exist.
follow-up: 12 comment:13 by , 9 years ago
Yeah, they took on board my suggestions for forcefield.py, but messed it up. In their version, the ignoreExternalBonds switch makes it ignore external bonds even in residues that are actually bonded, which is disastrous - disulfides, for example, end up with no angle, length, or torsion restraints on their S-S bonds. The correct thing to do is to check for the bonded topology first, and only re-do the check ignoring external bonds if the first check fails. But I guess the correct thing for me to do is to fix the official version of forcefield.py rather than sticking with my own.
comment:14 by , 9 years ago
7.1.1 should be substantially faster, indeed. They fixed the coordinate export function so that it can export directly as a Numpy array rather than as a list of objects that get converted in a Python loop. That's about 30ms per graphics update on the average system right there.
comment:15 by , 9 years ago
By the looks of the discussion at https://github.com/pandegroup/openmm/issues/1793, a Windows build should be coming soon. They're just hung up on a dependency issue due to some changes in the build system. I'll keep an eye on that and let you know when it becomes available.
comment:16 by , 9 years ago
If you need forcefield.py from OpenMM 7.1.1 patched in ChimeraX you will need to give me a patched version of that file. It has changed a lot since the 7.0.1 forcefield.py. As you say it is better to get the fix into OpenMM (7.1.2?) but perhaps that will not be ready in time for your workshop.
follow-up: 16 comment:17 by , 9 years ago
Tristan, you said you did not get email when I commented on this ticket. Did you get email for this comment? Eric enabled Trac logging so we can see what happened if you don’t receive this in email.
follow-up: 17 comment:18 by , 9 years ago
Looks like from the Trac log the previous comment was not emailed to Tristan. But it was emailed to goddard. That previous comment was submitted by me by email. This comment is being submitted directly in the web browser Trac interface.
comment:19 by , 9 years ago
Windows build is done: https://anaconda.org/omnia/openmm/7.1.1/download/win-64/openmm-7.1.1-py36_0.tar.bz2. On 2017-05-11 00:17, ChimeraX wrote:
follow-up: 19 comment:20 by , 9 years ago
I've updated to OpenMM 7.1.1 on Windows. I did not test it yet. Will wait to test tonight's daily build.
Also Eric Pettersen and Conrad Huang have supposedly fixed Trac so that comments on tickets will always be emailed to the owner and reporter and cc list.
comment:21 by , 9 years ago
My OpenMM Windows 7.1.1 in ChimeraX did not work (no platform "CPU"), but I fixed an OpenMM patch that tells it where to find its plugins and that worked. So Windows OpenMM should work in tonight's daily build.
comment:22 by , 9 years ago
| Owner: | changed from to |
|---|
comment:23 by , 9 years ago
Hi Tom, I've changed the new version of forcefield.py to work with ISOLDE. It will do the job for now, but in the longer run I want to do a fairly substantial overhaul of the simulation initialisation process. It looks like it will be possible to build a master OpenMM System for the entire structure up-front, and then just cull the necessary fragments from that for each simulation - so forcefield.py would only ever get used for that initial System generation. That's for later, though - right now I've got a few more important things to get done. I've asked the Erice people to download and install the most recent daily build of ChimeraX (which will be either today's or tomorrow's), and I'll be standardising my ISOLDE builds to work against that until after the demo is done. Targeted rotamers are working nicely now, and I should be able to get targeted secondary structures and register shifts working in time as well. Cheers, Tristan On 2017-05-10 01:25, Tom Goddard wrote:
follow-up: 23 comment:24 by , 9 years ago
Spoke too soon. That version still had a typo in it. This one works
(with a few tweaks needed in ISOLDE). The basic issue is that in some
cases (well, only one that I know of so far), if you allow missing
external bonds you can run into ambiguity. If you have a cysteine with a
bare sulfur, is that actually disulfide bonded or genuinely
deprotonated? OpenMM wants to know, because the parameters for each are
of course very different. So they've included a new argument in
createSystem, residueTemplates, which takes a {[OpenMM Residue]:
template_name} dict to allow strict template matching. So all cysteines
have to be double-checked while building the simulation topology. No big
deal, in the grand scheme of things.
On 2017-05-15 10:51, ChimeraX wrote:
comment:25 by , 9 years ago
I've patched ChimeraX OpenMM to use the forcefield-1.py attached to this ticket. Will be in tonight's builds.
follow-up: 24 comment:26 by , 8 years ago
| Resolution: | → fixed |
|---|---|
| Status: | assigned → closed |
The daily builds are now using OpenMM 7.3.
Is this problem specific to a new model/map you are trying?
Does the previous test case that worked continue to work reliably? If the previous case also now has problems them maybe it is something other than OpenMM, like an updated Linux opencl driver, or a subtle change to your own code, or a change to the ChimeraX code.
I guess you have checked old systems that it used to work on and it still works on those, so it is something special about the new system that triggers the bug. Please confirm that this guess is correct. If this is the case I guess the first thing to do is update to OpenMM 7.1. I can do that, but I'm going on vacation this weekend for 2 weeks and can't do it today. So if you want to pursue it sooner, you should simply install OpenMM 7.1 yourself. You can probably use the ChimeraX python to do the install so it is installed into ChimeraX.