#5536 closed defect (fixed)
Duplicate pseudobond depiction
| Reported by: | Owned by: | Greg Couch | |
|---|---|---|---|
| Priority: | normal | Milestone: | 1.3 |
| Component: | Graphics | Version: | |
| Keywords: | Cc: | Eric Pettersen, Tom Goddard | |
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
The following bug report has been submitted:
Platform: macOS-10.15.7-x86_64-i386-64bit
ChimeraX Version: 1.2.5 (2021-05-24 04:13:57 UTC)
Description
Pseudobond is generated twice: From atom to "cartoon end" and again from atom to (hidden) atom. Details and screenshots in attachment.
OpenGL version: 4.1 ATI-3.10.22
OpenGL renderer: AMD Radeon Pro 5500M OpenGL Engine
OpenGL vendor: ATI Technologies Inc.Hardware:
Hardware Overview:
Model Name: MacBook Pro
Model Identifier: MacBookPro16,1
Processor Name: 8-Core Intel Core i9
Processor Speed: 2.4 GHz
Number of Processors: 1
Total Number of Cores: 8
L2 Cache (per Core): 256 KB
L3 Cache: 16 MB
Hyper-Threading Technology: Enabled
Memory: 32 GB
Boot ROM Version: 1554.140.20.0.0 (iBridge: 18.16.14759.0.1,0)
Software:
System Software Overview:
System Version: macOS 10.15.7 (19H1323)
Kernel Version: Darwin 19.6.0
Time since boot: 35 days 8:21
Graphics/Displays:
Intel UHD Graphics 630:
Chipset Model: Intel UHD Graphics 630
Type: GPU
Bus: Built-In
VRAM (Dynamic, Max): 1536 MB
Vendor: Intel
Device ID: 0x3e9b
Revision ID: 0x0002
Automatic Graphics Switching: Supported
gMux Version: 5.0.0
Metal: Supported, feature set macOS GPUFamily2 v1
AMD Radeon Pro 5500M:
Chipset Model: AMD Radeon Pro 5500M
Type: GPU
Bus: PCIe
PCIe Lane Width: x8
VRAM (Total): 8 GB
Vendor: AMD (0x1002)
Device ID: 0x7340
Revision ID: 0x0040
ROM Revision: 113-D3220E-190
VBIOS Version: 113-D32206U1-020
Option ROM Version: 113-D32206U1-020
EFI Driver Version: 01.A1.190
Automatic Graphics Switching: Supported
gMux Version: 5.0.0
Metal: Supported, feature set macOS GPUFamily2 v1
Displays:
Color LCD:
Display Type: Built-In Retina LCD
Resolution: 3072 x 1920 Retina
Framebuffer Depth: 24-Bit Color (ARGB8888)
Main Display: Yes
Mirror: Off
Online: Yes
Automatically Adjust Brightness: No
Connection Type: Internal
Locale: (None, 'UTF-8')
PyQt5 5.15.2, Qt 5.15.2
Installed Packages:
alabaster: 0.7.12
appdirs: 1.4.4
appnope: 0.1.2
Babel: 2.9.1
backcall: 0.2.0
blockdiag: 2.0.1
certifi: 2020.12.5
cftime: 1.5.0
chardet: 3.0.4
ChimeraX-AddCharge: 1.0.1
ChimeraX-AddH: 2.1.6
ChimeraX-AlignmentAlgorithms: 2.0
ChimeraX-AlignmentHdrs: 3.2
ChimeraX-AlignmentMatrices: 2.0
ChimeraX-Alignments: 2.1
ChimeraX-AmberInfo: 1.0
ChimeraX-Arrays: 1.0
ChimeraX-Atomic: 1.13.2
ChimeraX-AtomicLibrary: 3.1.3
ChimeraX-AtomSearch: 2.0
ChimeraX-AtomSearchLibrary: 1.0
ChimeraX-AxesPlanes: 2.0
ChimeraX-BasicActions: 1.1
ChimeraX-BILD: 1.0
ChimeraX-BlastProtein: 1.1
ChimeraX-BondRot: 2.0
ChimeraX-BugReporter: 1.0
ChimeraX-BuildStructure: 2.5.2
ChimeraX-Bumps: 1.0
ChimeraX-BundleBuilder: 1.1
ChimeraX-ButtonPanel: 1.0
ChimeraX-CageBuilder: 1.0
ChimeraX-CellPack: 1.0
ChimeraX-Centroids: 1.1
ChimeraX-ChemGroup: 2.0
ChimeraX-Clashes: 2.1
ChimeraX-Clipper: 0.16.1
ChimeraX-ColorActions: 1.0
ChimeraX-ColorGlobe: 1.0
ChimeraX-ColorKey: 1.2.1
ChimeraX-CommandLine: 1.1.4
ChimeraX-ConnectStructure: 2.0
ChimeraX-Contacts: 1.0
ChimeraX-Core: 1.2.5
ChimeraX-CoreFormats: 1.0
ChimeraX-coulombic: 1.1.1
ChimeraX-Crosslinks: 1.0
ChimeraX-Crystal: 1.0
ChimeraX-CrystalContacts: 1.0
ChimeraX-DataFormats: 1.1
ChimeraX-Dicom: 1.0
ChimeraX-DistMonitor: 1.1.3
ChimeraX-DistUI: 1.0
ChimeraX-Dssp: 2.0
ChimeraX-EMDB-SFF: 1.0
ChimeraX-ExperimentalCommands: 1.0
ChimeraX-FileHistory: 1.0
ChimeraX-FunctionKey: 1.0
ChimeraX-Geometry: 1.1
ChimeraX-gltf: 1.0
ChimeraX-Graphics: 1.0
ChimeraX-Hbonds: 2.1
ChimeraX-Help: 1.1
ChimeraX-HKCage: 1.3
ChimeraX-IHM: 1.0
ChimeraX-ImageFormats: 1.1
ChimeraX-IMOD: 1.0
ChimeraX-IO: 1.0.1
ChimeraX-ISOLDE: 1.2.2
ChimeraX-Label: 1.0
ChimeraX-ListInfo: 1.1.1
ChimeraX-Log: 1.1.2
ChimeraX-LookingGlass: 1.1
ChimeraX-Maestro: 1.8.1
ChimeraX-Map: 1.0.2
ChimeraX-MapData: 2.0
ChimeraX-MapEraser: 1.0
ChimeraX-MapFilter: 2.0
ChimeraX-MapFit: 2.0
ChimeraX-MapSeries: 2.0
ChimeraX-Markers: 1.0
ChimeraX-Mask: 1.0
ChimeraX-MatchMaker: 1.2.1
ChimeraX-MDcrds: 2.2
ChimeraX-MedicalToolbar: 1.0.1
ChimeraX-Meeting: 1.0
ChimeraX-MLP: 1.1
ChimeraX-mmCIF: 2.3
ChimeraX-MMTF: 2.1
ChimeraX-Modeller: 1.0.1
ChimeraX-ModelPanel: 1.0.1
ChimeraX-ModelSeries: 1.0
ChimeraX-Mol2: 2.0
ChimeraX-Morph: 1.0
ChimeraX-MouseModes: 1.1
ChimeraX-Movie: 1.0
ChimeraX-Neuron: 1.0
ChimeraX-Nucleotides: 2.0.1
ChimeraX-OpenCommand: 1.5
ChimeraX-PDB: 2.4.1
ChimeraX-PDBBio: 1.0
ChimeraX-PDBLibrary: 1.0.1
ChimeraX-PDBMatrices: 1.0
ChimeraX-Phenix: 0.3
ChimeraX-PickBlobs: 1.0
ChimeraX-Positions: 1.0
ChimeraX-PresetMgr: 1.0.1
ChimeraX-PubChem: 2.0.1
ChimeraX-ReadPbonds: 1.0
ChimeraX-Registration: 1.1
ChimeraX-RemoteControl: 1.0
ChimeraX-ResidueFit: 1.0
ChimeraX-RestServer: 1.1
ChimeraX-RNALayout: 1.0
ChimeraX-RotamerLibMgr: 2.0
ChimeraX-RotamerLibsDunbrack: 2.0
ChimeraX-RotamerLibsDynameomics: 2.0
ChimeraX-RotamerLibsRichardson: 2.0
ChimeraX-SaveCommand: 1.4
ChimeraX-SchemeMgr: 1.0
ChimeraX-SDF: 2.0
ChimeraX-Segger: 1.0
ChimeraX-Segment: 1.0
ChimeraX-SeqView: 2.3
ChimeraX-Shape: 1.0.1
ChimeraX-Shell: 1.0
ChimeraX-Shortcuts: 1.0
ChimeraX-ShowAttr: 1.0
ChimeraX-ShowSequences: 1.0
ChimeraX-SideView: 1.0
ChimeraX-Smiles: 2.0.1
ChimeraX-SmoothLines: 1.0
ChimeraX-SpaceNavigator: 1.0
ChimeraX-StdCommands: 1.3.1
ChimeraX-STL: 1.0
ChimeraX-Storm: 1.0
ChimeraX-Struts: 1.0
ChimeraX-Surface: 1.0
ChimeraX-SwapAA: 2.0
ChimeraX-SwapRes: 2.1
ChimeraX-TapeMeasure: 1.0
ChimeraX-Test: 1.0
ChimeraX-Toolbar: 1.0.1
ChimeraX-ToolshedUtils: 1.2
ChimeraX-Tug: 1.0
ChimeraX-UI: 1.7.6
ChimeraX-uniprot: 2.1
ChimeraX-UnitCell: 1.0
ChimeraX-ViewDockX: 1.0
ChimeraX-Vive: 1.1
ChimeraX-VolumeMenu: 1.0
ChimeraX-VTK: 1.0
ChimeraX-WavefrontOBJ: 1.0
ChimeraX-WebCam: 1.0
ChimeraX-WebServices: 1.0
ChimeraX-Zone: 1.0
colorama: 0.4.3
comtypes: 1.1.7
cxservices: 1.0
cycler: 0.10.0
Cython: 0.29.21
decorator: 5.0.9
distlib: 0.3.1
docutils: 0.16
filelock: 3.0.12
funcparserlib: 0.3.6
grako: 3.16.5
h5py: 2.10.0
html2text: 2020.1.16
idna: 2.10
ihm: 0.17
imagecodecs: 2020.5.30
imagesize: 1.2.0
ipykernel: 5.3.4
ipython: 7.18.1
ipython-genutils: 0.2.0
jedi: 0.17.2
Jinja2: 2.11.2
jupyter-client: 6.1.7
jupyter-core: 4.7.1
kiwisolver: 1.3.1
line-profiler: 2.1.2
lxml: 4.6.2
lz4: 3.1.0
MarkupSafe: 2.0.1
matplotlib: 3.3.2
matplotlib-inline: 0.1.2
msgpack: 1.0.0
netCDF4: 1.5.4
networkx: 2.5
numexpr: 2.7.3
numpy: 1.19.2
numpydoc: 1.1.0
openvr: 1.14.1501
packaging: 20.9
ParmEd: 3.2.0
parso: 0.7.1
pexpect: 4.8.0
pickleshare: 0.7.5
Pillow: 7.2.0
pip: 21.0.1
pkginfo: 1.5.0.1
prompt-toolkit: 3.0.18
psutil: 5.7.2
ptyprocess: 0.7.0
pycollada: 0.7.1
pydicom: 2.0.0
Pygments: 2.7.1
PyOpenGL: 3.1.5
PyOpenGL-accelerate: 3.1.5
pyparsing: 2.4.7
PyQt5-commercial: 5.15.2
PyQt5-sip: 12.8.1
PyQtWebEngine-commercial: 5.15.2
python-dateutil: 2.8.1
pytz: 2021.1
pyzmq: 22.0.3
qtconsole: 4.7.7
QtPy: 1.9.0
RandomWords: 0.3.0
requests: 2.24.0
scipy: 1.5.2
Send2Trash: 1.5.0
SEQCROW: 1.3
setuptools: 50.3.2
sfftk-rw: 0.6.7.dev1
six: 1.15.0
snowballstemmer: 2.1.0
sortedcontainers: 2.2.2
Sphinx: 3.2.1
sphinxcontrib-applehelp: 1.0.2
sphinxcontrib-blockdiag: 2.0.0
sphinxcontrib-devhelp: 1.0.2
sphinxcontrib-htmlhelp: 2.0.0
sphinxcontrib-jsmath: 1.0.1
sphinxcontrib-qthelp: 1.0.3
sphinxcontrib-serializinghtml: 1.1.5
suds-jurko: 0.6
tables: 3.6.1
tifffile: 2020.9.3
tinyarray: 1.2.3
tornado: 6.1
traitlets: 5.0.5
urllib3: 1.25.11
wcwidth: 0.2.5
webcolors: 1.11.1
wheel: 0.36.0
wheel-filename: 1.3.0
File attachment: ChimeraX_pbond_bug_report.png
{kind=link}
{kind=link}
Attachments (2)
Change History (15)
by , 5 years ago
| Attachment: | ChimeraX_pbond_bug_report.png added |
|---|
comment:1 by , 5 years ago
| Cc: | added |
|---|---|
| Component: | Unassigned → Graphics |
| Owner: | set to |
| Platform: | → all |
| Project: | → ChimeraX |
| Status: | new → assigned |
| Summary: | ChimeraX bug report submission → Duplicate pseudobond depiction |
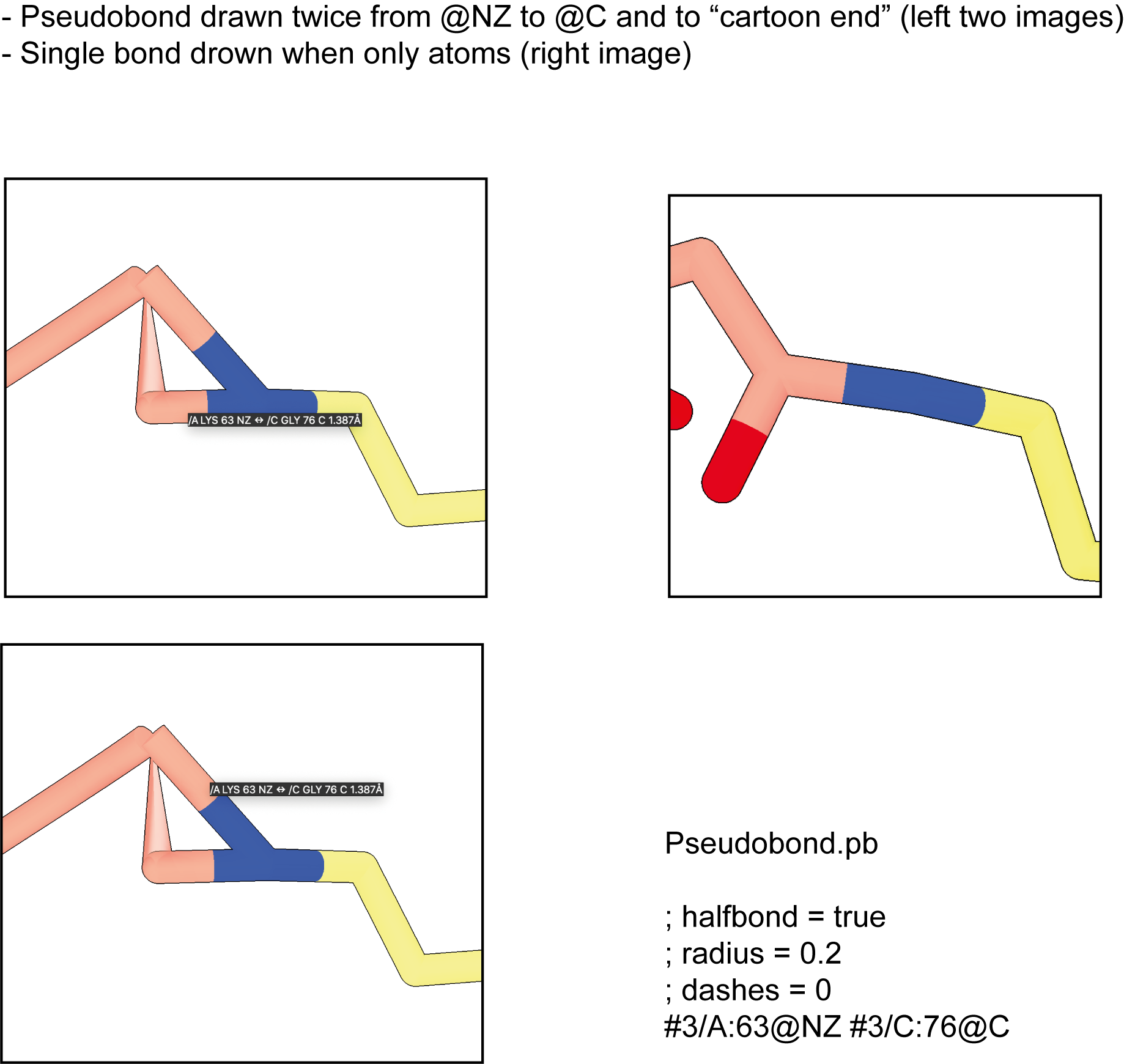
Reported by Sven Lange
comment:2 by , 5 years ago
I see you made a pseudobond from G76C to L63N. The weird transparent cone you see is not a pseudobond, that is a tether meant to connect a sidechain to a ribbon, typically drawn between the CA atom and the actual ribbon position which does not exactly go through the CA atom position. When you show ribbon your pseudobond seems to be going to the ribbon instead of to the C atoms. Pseudobonds do go to the ribbon and not the atom position when the atom is not displayed and the ribbon is shown -- they are projected onto the ribbon because otherwise the pseudobond ends in space on no visible atoms which is confusing. But in your case you have the C atom shown so it should not go to the ribbon. I'll try this to see if I can understand what is going on. We fixed a bug like this a month ago related to atom positions on ribbons. You might try the ChimeraX 1.3 release candidate since the ChimeraX 1.2.5 version you are using does not have that fix.
comment:3 by , 5 years ago
I am baffled how you got two pseudobonds shown, one to the ribbon of residue 76 and one to the C atom of residue 76. Also I guess you turned off suppressing ribbon backbone atom display (cartoon #3 suppress false) in order to see the C backbone atom of residue 76 which would normally be hidden when the ribbon is shown.
Tests in ChimeraX 1.2.5 and the ChimeraX 1.3 release candidate show that 1.2.5 which you are using has a bug where the pseudobond is drawn to the ribbon even when the C atom is shown while ChimeraX 1.3 does not have that bug. So I suggest trying ChimeraX 1.3 release candidate.
If that does not fix the problem then please attach a ChimeraX session that illustrates this problem. I can't debug it with just a picture and not even the commands that produced that picture.
comment:4 by , 5 years ago
Hi Tom,
Thanks for your reply and sorry I didn’t follow up on this earlier, I just found these emails in my spam folder!
1. The duplicate pseudobond problem persists in ChimeraX 1.3.
2. Interestingly, the problem occurs with phenix.refine output pdb files only, not with structures fetched from the PDB.
I have attached an example of the same structure where the pbonds are shown correctly when opened from the PDB, but bonds show incorrectly when using the phenix.refine output pdb (same structure prior to PDB deposition).
The duplicate pbond problem is reproducible among all (3) different structures that I have tried it on when using the phenix.refine output pdb.
My workaround for the structures that are not available from the PDB yet is to draw a bond and then show the tether as cylinder, however, this can lead to quite ugly ‘bond angles’. E.g. pseudobond between K63NZ and G76C:
color /A:63@NZ /C:76@C halfbond true
cartoon tether shape cylinder opacity 1
I hope the attached scripts help to troubleshoot this, happy to discuss further!
BW
Sven
From: ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu>
Date: Friday, 5 November 2021 at 02:38
To:
Cc: goddard@cgl.ucsf.edu <goddard@cgl.ucsf.edu>, pett@cgl.ucsf.edu <pett@cgl.ucsf.edu>, Sven Lange (Staff) <SLange001@dundee.ac.uk>
Subject: Re: [ChimeraX] #5536: Duplicate pseudobond depiction
CAUTION: This email originated from outside the University of Dundee. Do not click links or open attachments unless you recognise the sender's email address and know the content is safe.
#5536: Duplicate pseudobond depiction
----------------------------------+-------------------------
Reporter: slange001@… | Owner: Tom Goddard
Type: defect | Status: assigned
Priority: normal | Milestone:
Component: Graphics | Version:
Resolution: | Keywords:
Blocked By: | Blocking:
Notify when closed: | Platform: all
Project: ChimeraX |
----------------------------------+-------------------------
Comment (by Tom Goddard):
I am baffled how you got two pseudobonds shown, one to the ribbon of
residue 76 and one to the C atom of residue 76. Also I guess you turned
off suppressing ribbon backbone atom display (cartoon #3 suppress false)
in order to see the C backbone atom of residue 76 which would normally be
hidden when the ribbon is shown.
Tests in ChimeraX 1.2.5 and the ChimeraX 1.3 release candidate show that
1.2.5 which you are using has a bug where the pseudobond is drawn to the
ribbon even when the C atom is shown while ChimeraX 1.3 does not have that
bug. So I suggest trying ChimeraX 1.3 release candidate.
If that does not fix the problem then please attach a ChimeraX session
that illustrates this problem. I can't debug it with just a picture and
not even the commands that produced that picture.
--
Ticket URL: <https://www.rbvi.ucsf.edu/trac/ChimeraX/ticket/5536#comment:3>
ChimeraX <https://www.rbvi.ucsf.edu/chimerax/>
ChimeraX Issue Tracker
The University of Dundee is a registered Scottish Charity, No: SC015096
follow-up: 4 comment:5 by , 5 years ago
It seems that the M2Ub5_phenix_pseudobonds.pb file is creating 6 pseudobonds ("Opened Pseudobonds M2Ub5_phenix_pseudobonds.pb, 6 bonds") when it should only create 3 AFAICT.
comment:6 by , 5 years ago
Yes, I was wondering what that number means… 1. 6 bonds are created in both cases (phenix.refine and PDB file), but ChimeraX only displays 6 for the phenix.refine file. 2. In the phenix.refine script, if I delete all three specified bonds from the pseudobond file (only leave the first three lines of the file as they are), it still reports 4 bonds created and displays only the three pseudobonds to the tethered C atoms, but not to the ribbon. The University of Dundee is a registered Scottish Charity, No: SC015096
comment:7 by , 5 years ago
Okay, the reported number of pseudobonds was a red herring -- that code was counting the total number of lines in the file and not accounting for the control lines. I have committed a fix for that. At any rate, only 3 pseudobonds are actually created.
follow-up: 6 comment:8 by , 5 years ago
Ah, that’s it. So this would mean that the extra unwanted pbonds to the atom C are already pre-existing - And the script only creates the three pbonds to the ribbon C. It seems that the LINK entries interpreted as pbonds. The phenix.refine file contains these entries (I defined the bonds in phenix.refine to keep the atoms close during refinement) - the deposited PDB entry does not contain them. LINK NZ LYS B 48 C GLY C 76 LINK NZ LYS C 48 C GLY D 76 LINK NZ LYS E 48 C GLY F 76 When I delete these from the phenix.refine file, the unwanted pbonds disappear too. The University of Dundee is a registered Scottish Charity, No: SC015096
comment:9 by , 5 years ago
Yes, the PDB reader creates covalent (not pseudo) bonds for those LINK records. What you reported is still a bug in that the covalent bond causes the C atom to be displayed with a tether to the ribbon, and the pseudobond should go to that depicted C instead of straight to the ribbon. It should only go to the ribbon if the C is not explicitly depicted. So that is a bug,
Still, it seems that you have a workaround for your purposes.
follow-up: 8 comment:10 by , 5 years ago
Thanks for your help - and for developing such amazing software! The University of Dundee is a registered Scottish Charity, No: SC015096
comment:11 by , 5 years ago
| Cc: | added |
|---|---|
| Milestone: | → 1.3 |
| Owner: | changed from to |
Fixed in ChimeraX 1.3 release candidate and 1.4 daily builds.
The Atom.effective_coord() C++ method for some reason gave the ribbon coordinate when the atom was displayed. It is only used for pseudobond display and by nucleotides display. I have changed it so it returns the atom coordinate if the atom is shown as I believe that is the intended behavior. There is a separate Atom.ribbon_coord() that always gives the ribbon coordinate.
I am not sure if this change is the correct behavior for the nucleotides depiction code use of Atom.effective_coord(). Reassigning to Greg to assess whether nucleotides needs fixing.
follow-up: 10 comment:12 by , 5 years ago
| Resolution: | → fixed |
|---|---|
| Status: | assigned → closed |
Nucleotides deliberately uses Atom.effective_coord() instead of Atom.ribbon_coord().
comment:13 by , 5 years ago
This pseudobond fix was not correct and caused a new problem #5732 which provided the correct fix.
Added by email2trac