#5045 closed defect (fixed)
Modeller sequence mismatch failure
| Reported by: | Owned by: | Eric Pettersen | |
|---|---|---|---|
| Priority: | normal | Milestone: | |
| Component: | Sequence | Version: | |
| Keywords: | Cc: | ||
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
The following bug report has been submitted:
Platform: Windows-10-10.0.19041
ChimeraX Version: 1.2.5 (2021-05-24 04:13:57 UTC)
Description
I like to build a multichain homology model using Modeller Comparative, but was unable to get output model. The template structure PDB number is 4n9f. The sequence alignment of target sequence and template sequence are open (see attached). The error is shown below:
No output models from Modeller; see log for Modeller text output.
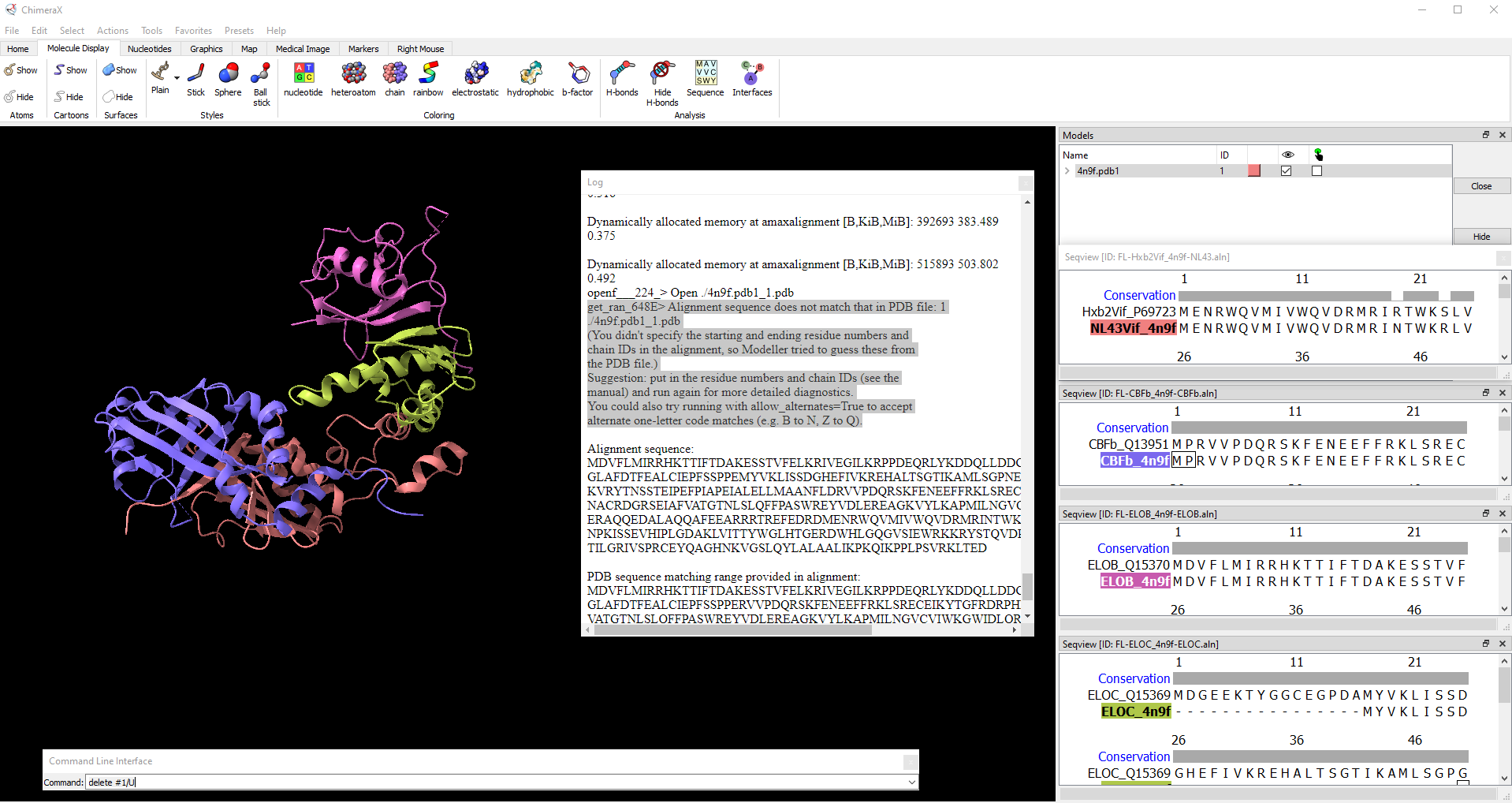
Alignment sequence does not match that in PDB file: 1 ./4n9f.pdb1_1.pdb
(You didn't specify the starting and ending residue numbers and
chain IDs in the alignment, so Modeller tried to guess these from
the PDB file.)
Suggestion: put in the residue numbers and chain IDs (see the
manual) and run again for more detailed diagnostics.
You could also try running with allow_alternates=True to accept
alternate one-letter code matches (e.g. B to N, Z to Q).
Already try many hours on it but still failed to figure out how to fix it. Could you please give me some instruction? Thank you
Log:
UCSF ChimeraX version: 1.2.5 (2021-05-24)
© 2016-2021 Regents of the University of California. All rights reserved.
How to cite UCSF ChimeraX
> open "E:\temp\EM data\4n9f.pdb1" format pdb
Summary of feedback from opening E:\temp\EM data\4n9f.pdb1
---
warnings | Start residue of secondary structure not found: HELIX 35 35 LYS C
18 GLN C 32 1 15
Start residue of secondary structure not found: HELIX 36 36 THR C 36 ASP C 54
1 19
Start residue of secondary structure not found: HELIX 37 37 LYS C 56 SER C 82
1 27
Start residue of secondary structure not found: HELIX 38 38 ASP C 85 LEU C 107
1 23
Start residue of secondary structure not found: HELIX 39 39 PRO C 108 PRO C
110 5 3
680 messages similar to the above omitted
Cannot find LINK/SSBOND residue HIS (139 )
Cannot find LINK/SSBOND residue HIS (139 )
Cannot find LINK/SSBOND residue CYS (114 )
Cannot find LINK/SSBOND residue CYS (114 )
Cannot find LINK/SSBOND residue CYS (133 )
33 messages similar to the above omitted
4n9f.pdb1 title:
Crystal structure of the vif-cbfbeta-CUL5-elob-eloc pentameric complex [more
info...]
Chain information for 4n9f.pdb1 #1
---
Chain | Description
U | No description available
X | No description available
Y | No description available
a | No description available
b | No description available
Non-standard residues in 4n9f.pdb1 #1
---
ZN — zinc ion
> hide atoms
> show cartoons
> delete #1/U
> open "E:/temp/EM data/FL-Hxb2Vif_4n9f-NL43.aln"
Summary of feedback from opening E:/temp/EM data/FL-Hxb2Vif_4n9f-NL43.aln
---
notes | Alignment identifier is FL-Hxb2Vif_4n9f-NL43.aln
Associated 4n9f.pdb1 chain b to NL43Vif_4n9f with 0 mismatches
Showing conservation header ("seq_conservation" residue attribute) for
alignment FL-Hxb2Vif_4n9f-NL43.aln
Opened 2 sequences from FL-Hxb2Vif_4n9f-NL43.aln
> sequence associate /b FL-Hxb2Vif_4n9f-NL43.aln:1
Disassociated 4n9f.pdb1 chain b from NL43Vif_4n9f
Associated 4n9f.pdb1 chain b to Hxb2Vif_P69723 with 20 mismatches and/or gaps
> sequence associate /b FL-Hxb2Vif_4n9f-NL43.aln:2
Disassociated 4n9f.pdb1 chain b from Hxb2Vif_P69723
Associated 4n9f.pdb1 chain b to NL43Vif_4n9f with 0 mismatches
> sequence associate /b FL-Hxb2Vif_4n9f-NL43.aln:1
Disassociated 4n9f.pdb1 chain b from NL43Vif_4n9f
Associated 4n9f.pdb1 chain b to Hxb2Vif_P69723 with 20 mismatches and/or gaps
> sequence associate /b FL-Hxb2Vif_4n9f-NL43.aln:2
Disassociated 4n9f.pdb1 chain b from Hxb2Vif_P69723
Associated 4n9f.pdb1 chain b to NL43Vif_4n9f with 0 mismatches
> sequence associate /b FL-Hxb2Vif_4n9f-NL43.aln:1
Disassociated 4n9f.pdb1 chain b from NL43Vif_4n9f
Associated 4n9f.pdb1 chain b to Hxb2Vif_P69723 with 20 mismatches and/or gaps
> open "E:/temp/EM data/FL-CBFb_4n9f-CBFb.aln" "E:/temp/EM data/FL-
> ELOB_4n9f-ELOB.aln" "E:/temp/EM data/FL-ELOC_4n9f-ELOC.aln"
Summary of feedback from opening E:/temp/EM data/FL-CBFb_4n9f-CBFb.aln
---
notes | Alignment identifier is FL-CBFb_4n9f-CBFb.aln
Associated 4n9f.pdb1 chain a to CBFb_4n9f with 0 mismatches
Showing conservation header ("seq_conservation" residue attribute) for
alignment FL-CBFb_4n9f-CBFb.aln
Summary of feedback from opening E:/temp/EM data/FL-ELOB_4n9f-ELOB.aln
---
notes | Alignment identifier is FL-ELOB_4n9f-ELOB.aln
Associated 4n9f.pdb1 chain X to ELOB_4n9f with 0 mismatches
Showing conservation header ("seq_conservation" residue attribute) for
alignment FL-ELOB_4n9f-ELOB.aln
Summary of feedback from opening E:/temp/EM data/FL-ELOC_4n9f-ELOC.aln
---
notes | Alignment identifier is FL-ELOC_4n9f-ELOC.aln
Associated 4n9f.pdb1 chain Y to ELOC_4n9f with 0 mismatches
Showing conservation header ("seq_conservation" residue attribute) for
alignment FL-ELOC_4n9f-ELOC.aln
Opened 2 sequences from FL-CBFb_4n9f-CBFb.aln
Opened 2 sequences from FL-ELOB_4n9f-ELOB.aln
Opened 2 sequences from FL-ELOC_4n9f-ELOC.aln
> sequence associate /X FL-ELOB_4n9f-ELOB.aln:1
Disassociated 4n9f.pdb1 chain X from ELOB_4n9f
Associated 4n9f.pdb1 chain X to ELOB_Q15370 with 0 mismatches
> select /a:5
7 atoms, 6 bonds, 1 residue, 1 model selected
> select /a:5
7 atoms, 6 bonds, 1 residue, 1 model selected
> sequence associate /a FL-CBFb_4n9f-CBFb.aln:1
Disassociated 4n9f.pdb1 chain a from CBFb_4n9f
Associated 4n9f.pdb1 chain a to CBFb_Q13951 with 0 mismatches
> sequence associate /Y FL-ELOC_4n9f-ELOC.aln:1
Disassociated 4n9f.pdb1 chain Y from ELOC_4n9f
Associated 4n9f.pdb1 chain Y to ELOC_Q15369 with 0 mismatches
> ui tool show "Modeller Comparative"
> modeller comparative FL-CBFb_4n9f-CBFb.aln:1 FL-ELOB_4n9f-ELOB.aln:1 FL-
> ELOC_4n9f-ELOC.aln:1 FL-Hxb2Vif_4n9f-NL43.aln:1 multichain true numModels 5
> fast false hetPreserve false hydrogens false waterPreserve false
Web Service: Modeller9v8 is a Python wrapper that calls Modeller (v9.18) for
protein structure modeling
Opal service URL:
http://webservices.rbvi.ucsf.edu/opal2/services/Modeller9v8Service
Opal job id: appModeller9v8Service1628754749123800597657
Opal status URL prefix:
http://webservices.rbvi.ucsf.edu/appModeller9v8Service1628754749123800597657
stdout.txt = standard output
stderr.txt = standard error
Modeller job ID appModeller9v8Service1628754749123800597657 finished
Modeller error output
Traceback (most recent call last):
File "/usr/local/opal-local/bin/modeller9v8.py", line 322, in <module>
main()
File "/usr/local/opal-local/bin/modeller9v8.py", line 24, in main
VersionMap[cf["version"]](cf)
File "/usr/local/opal-local/bin/modeller9v8.py", line 34, in v2_run
execfile(fn)
File "ModellerModelling.py", line 67, in <module>
a.make()
File "/usr/lib64/python2.7/site-packages/modeller/automodel/automodel.py",
line 133, in make
self.homcsr(exit_stage)
File "/usr/lib64/python2.7/site-packages/modeller/automodel/automodel.py",
line 595, in homcsr
aln = self.read_alignment()
File "/usr/lib64/python2.7/site-packages/modeller/automodel/automodel.py",
line 556, in read_alignment
aln.append(file=self.alnfile, align_codes=codes)
File "/usr/lib64/python2.7/site-packages/modeller/alignment.py", line 80, in
append
allow_alternates)
_modeller.SequenceMismatchError: get_ran_648E> Alignment sequence does not
match that in PDB file: 1 ./4n9f.pdb1_1.pdb (You didn't specify the starting
and ending residue numbers and chain IDs in the alignment, so Modeller tried
to guess these from the PDB file.) Suggestion: put in the residue numbers and
chain IDs (see the manual) and run again for more detailed diagnostics. You
could also try running with allow_alternates=True to accept alternate one-
letter code matches (e.g. B to N, Z to Q).
Modeller run output
MODELLER 9.22, 2019/06/19, r11413
PROTEIN STRUCTURE MODELLING BY SATISFACTION OF SPATIAL RESTRAINTS
Copyright(c) 1989-2019 Andrej Sali
All Rights Reserved
Written by A. Sali
with help from
B. Webb, M.S. Madhusudhan, M-Y. Shen, G.Q. Dong,
M.A. Marti-Renom, N. Eswar, F. Alber, M. Topf, B. Oliva,
A. Fiser, R. Sanchez, B. Yerkovich, A. Badretdinov,
F. Melo, J.P. Overington, E. Feyfant
University of California, San Francisco, USA
Rockefeller University, New York, USA
Harvard University, Cambridge, USA
Imperial Cancer Research Fund, London, UK
Birkbeck College, University of London, London, UK
Kind, OS, HostName, Kernel, Processor: 4, Linux franklin.cgl.ucsf.edu
3.10.0-1160.36.2.el7.x86_64 x86_64
Date and time of compilation : 2019/06/19 14:00:54
MODELLER executable type : x86_64-intel8
Job starting time (YY/MM/DD HH:MM:SS): 2021/08/12 00:52:29
openf___224_> Open $(LIB)/restyp.lib
openf___224_> Open ${MODINSTALL9v22}/modlib/resgrp.lib
rdresgr_266_> Number of residue groups: 2
openf___224_> Open ${MODINSTALL9v22}/modlib/sstruc.lib
Dynamically allocated memory at amaxlibraries [B,KiB,MiB]: 191566 187.076
0.183
Dynamically allocated memory at amaxlibraries [B,KiB,MiB]: 192094 187.592
0.183
openf___224_> Open ${MODINSTALL9v22}/modlib/resdih.lib
Dynamically allocated memory at amaxlibraries [B,KiB,MiB]: 240694 235.053
0.230
rdrdih__263_> Number of dihedral angle types : 9
Maximal number of dihedral angle optima: 3
Dihedral angle names : Alph Phi Psi Omeg chi1 chi2 chi3 chi4 chi5
openf___224_> Open ${MODINSTALL9v22}/modlib/radii.lib
Dynamically allocated memory at amaxlibraries [B,KiB,MiB]: 253994 248.041
0.242
openf___224_> Open ${MODINSTALL9v22}/modlib/radii14.lib
openf___224_> Open ${MODINSTALL9v22}/modlib/af_mnchdef.lib
rdwilmo_274_> Mainchain residue conformation classes: APBLE
openf___224_> Open ${MODINSTALL9v22}/modlib/mnch.lib
rdclass_257_> Number of classes: 5
openf___224_> Open ${MODINSTALL9v22}/modlib/mnch1.lib
openf___224_> Open ${MODINSTALL9v22}/modlib/mnch2.lib
openf___224_> Open ${MODINSTALL9v22}/modlib/mnch3.lib
openf___224_> Open ${MODINSTALL9v22}/modlib/xs4.mat
rdrrwgh_268_> Number of residue types: 21
openf___224_> Open alignment.ali
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 269801 263.478
0.257
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 284893 278.216
0.272
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 300293 293.255
0.286
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 331093 323.333
0.316
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 392693 383.489
0.375
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 515893 503.802
0.492
openf___224_> Open ./4n9f.pdb1_1.pdb
get_ran_648E> Alignment sequence does not match that in PDB file: 1
./4n9f.pdb1_1.pdb
(You didn't specify the starting and ending residue numbers and
chain IDs in the alignment, so Modeller tried to guess these from
the PDB file.)
Suggestion: put in the residue numbers and chain IDs (see the
manual) and run again for more detailed diagnostics.
You could also try running with allow_alternates=True to accept
alternate one-letter code matches (e.g. B to N, Z to Q).
Alignment sequence:
MDVFLMIRRHKTTIFTDAKESSTVFELKRIVEGILKRPPDEQRLYKDDQLLDDGKTLGECGFTSQTARPQAPATV
GLAFDTFEALCIEPFSSPPEMYVKLISSDGHEFIVKREHALTSGTIKAMLSGPNEVNFREIPSHVLSKVCMYFTY
KVRYTNSSTEIPEFPIAPEIALELLMAANFLDRVVPDQRSKFENEEFFRKLSRECEIKYTGFRDRPHEERQARFQ
NACRDGRSEIAFVATGTNLSLQFFPASWREYVDLEREAGKVYLKAPMILNGVCVIWKGWIDLQRLDGMGCLEFDE
ERAQQEDALAQQAFEEARRRTREFEDRDMENRWQVMIVWQVDRMRINTWKRLVKHHMYISRKAKDWFYRHHYEST
NPKISSEVHIPLGDAKLVITTYWGLHTGERDWHLGQGVSIEWRKKRYSTQVDPDLADQLIHLHYFDCFSESAIRN
TILGRIVSPRCEYQAGHNKVGSLQYLALAALIKPKQIKPPLPSVRKLTED
PDB sequence matching range provided in alignment:
MDVFLMIRRHKTTIFTDAKESSTVFELKRIVEGILKRPPDEQRLYKDDQLLDDGKTLGECGFTSQTARPQAPATV
GLAFDTFEALCIEPFSSPPERVVPDQRSKFENEEFFRKLSRECEIKYTGFRDRPHEERQARFQNACRDGRSEIAF
VATGTNLSLQFFPASWREYVDLEREAGKVYLKAPMILNGVCVIWKGWIDLQRLDGMGCLEFDEERAQQEDALAQQ
AFEEARRRTREFEDRDMENRWQVMIVWQVDRMRINTWKRLVKHHMYISRKAKDWFYRHHYESTNPKISSEVHIPL
GDAKLVITTYWGLHTGERDWHLGQGVSIEWRKKRYSTQVDPDLADQLIHLHYFDCFSESAIRNTILGRIVSPRCE
YQAGHNKVGSLQYLALAALIKPKQIKPPLPSVRKLTED
No output models from Modeller; see log for Modeller text output.
> sequence associate /b FL-Hxb2Vif_4n9f-NL43.aln:2
Disassociated 4n9f.pdb1 chain b from Hxb2Vif_P69723
Associated 4n9f.pdb1 chain b to NL43Vif_4n9f with 0 mismatches
> sequence associate /a FL-CBFb_4n9f-CBFb.aln:2
Disassociated 4n9f.pdb1 chain a from CBFb_Q13951
Associated 4n9f.pdb1 chain a to CBFb_4n9f with 0 mismatches
> sequence associate /X FL-ELOB_4n9f-ELOB.aln:2
Disassociated 4n9f.pdb1 chain X from ELOB_Q15370
Associated 4n9f.pdb1 chain X to ELOB_4n9f with 0 mismatches
> sequence associate /Y FL-ELOC_4n9f-ELOC.aln:2
Disassociated 4n9f.pdb1 chain Y from ELOC_Q15369
Associated 4n9f.pdb1 chain Y to ELOC_4n9f with 0 mismatches
> ui tool show "Modeller Comparative"
> modeller comparative FL-CBFb_4n9f-CBFb.aln:1 FL-ELOB_4n9f-ELOB.aln:1 FL-
> ELOC_4n9f-ELOC.aln:1 FL-Hxb2Vif_4n9f-NL43.aln:1 multichain true numModels 5
> fast false hetPreserve false hydrogens false waterPreserve false
Web Service: Modeller9v8 is a Python wrapper that calls Modeller (v9.18) for
protein structure modeling
Opal service URL:
http://webservices.rbvi.ucsf.edu/opal2/services/Modeller9v8Service
Opal job id: appModeller9v8Service16287548655501857557318
Opal status URL prefix:
http://webservices.rbvi.ucsf.edu/appModeller9v8Service16287548655501857557318
stdout.txt = standard output
stderr.txt = standard error
Modeller job ID appModeller9v8Service16287548655501857557318 finished
Modeller error output
Traceback (most recent call last):
File "/usr/local/opal-local/bin/modeller9v8.py", line 322, in <module>
main()
File "/usr/local/opal-local/bin/modeller9v8.py", line 24, in main
VersionMap[cf["version"]](cf)
File "/usr/local/opal-local/bin/modeller9v8.py", line 34, in v2_run
execfile(fn)
File "ModellerModelling.py", line 67, in <module>
a.make()
File "/usr/lib64/python2.7/site-packages/modeller/automodel/automodel.py",
line 133, in make
self.homcsr(exit_stage)
File "/usr/lib64/python2.7/site-packages/modeller/automodel/automodel.py",
line 595, in homcsr
aln = self.read_alignment()
File "/usr/lib64/python2.7/site-packages/modeller/automodel/automodel.py",
line 556, in read_alignment
aln.append(file=self.alnfile, align_codes=codes)
File "/usr/lib64/python2.7/site-packages/modeller/alignment.py", line 80, in
append
allow_alternates)
_modeller.SequenceMismatchError: get_ran_648E> Alignment sequence does not
match that in PDB file: 1 ./4n9f.pdb1_1.pdb (You didn't specify the starting
and ending residue numbers and chain IDs in the alignment, so Modeller tried
to guess these from the PDB file.) Suggestion: put in the residue numbers and
chain IDs (see the manual) and run again for more detailed diagnostics. You
could also try running with allow_alternates=True to accept alternate one-
letter code matches (e.g. B to N, Z to Q).
Modeller run output
MODELLER 9.22, 2019/06/19, r11413
PROTEIN STRUCTURE MODELLING BY SATISFACTION OF SPATIAL RESTRAINTS
Copyright(c) 1989-2019 Andrej Sali
All Rights Reserved
Written by A. Sali
with help from
B. Webb, M.S. Madhusudhan, M-Y. Shen, G.Q. Dong,
M.A. Marti-Renom, N. Eswar, F. Alber, M. Topf, B. Oliva,
A. Fiser, R. Sanchez, B. Yerkovich, A. Badretdinov,
F. Melo, J.P. Overington, E. Feyfant
University of California, San Francisco, USA
Rockefeller University, New York, USA
Harvard University, Cambridge, USA
Imperial Cancer Research Fund, London, UK
Birkbeck College, University of London, London, UK
Kind, OS, HostName, Kernel, Processor: 4, Linux franklin.cgl.ucsf.edu
3.10.0-1160.36.2.el7.x86_64 x86_64
Date and time of compilation : 2019/06/19 14:00:54
MODELLER executable type : x86_64-intel8
Job starting time (YY/MM/DD HH:MM:SS): 2021/08/12 00:54:25
openf___224_> Open $(LIB)/restyp.lib
openf___224_> Open ${MODINSTALL9v22}/modlib/resgrp.lib
rdresgr_266_> Number of residue groups: 2
openf___224_> Open ${MODINSTALL9v22}/modlib/sstruc.lib
Dynamically allocated memory at amaxlibraries [B,KiB,MiB]: 191566 187.076
0.183
Dynamically allocated memory at amaxlibraries [B,KiB,MiB]: 192094 187.592
0.183
openf___224_> Open ${MODINSTALL9v22}/modlib/resdih.lib
Dynamically allocated memory at amaxlibraries [B,KiB,MiB]: 240694 235.053
0.230
rdrdih__263_> Number of dihedral angle types : 9
Maximal number of dihedral angle optima: 3
Dihedral angle names : Alph Phi Psi Omeg chi1 chi2 chi3 chi4 chi5
openf___224_> Open ${MODINSTALL9v22}/modlib/radii.lib
Dynamically allocated memory at amaxlibraries [B,KiB,MiB]: 253994 248.041
0.242
openf___224_> Open ${MODINSTALL9v22}/modlib/radii14.lib
openf___224_> Open ${MODINSTALL9v22}/modlib/af_mnchdef.lib
rdwilmo_274_> Mainchain residue conformation classes: APBLE
openf___224_> Open ${MODINSTALL9v22}/modlib/mnch.lib
rdclass_257_> Number of classes: 5
openf___224_> Open ${MODINSTALL9v22}/modlib/mnch1.lib
openf___224_> Open ${MODINSTALL9v22}/modlib/mnch2.lib
openf___224_> Open ${MODINSTALL9v22}/modlib/mnch3.lib
openf___224_> Open ${MODINSTALL9v22}/modlib/xs4.mat
rdrrwgh_268_> Number of residue types: 21
openf___224_> Open alignment.ali
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 269801 263.478
0.257
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 284893 278.216
0.272
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 300293 293.255
0.286
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 331093 323.333
0.316
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 392693 383.489
0.375
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 515893 503.802
0.492
openf___224_> Open ./4n9f.pdb1_1.pdb
get_ran_648E> Alignment sequence does not match that in PDB file: 1
./4n9f.pdb1_1.pdb
(You didn't specify the starting and ending residue numbers and
chain IDs in the alignment, so Modeller tried to guess these from
the PDB file.)
Suggestion: put in the residue numbers and chain IDs (see the
manual) and run again for more detailed diagnostics.
You could also try running with allow_alternates=True to accept
alternate one-letter code matches (e.g. B to N, Z to Q).
Alignment sequence:
MDVFLMIRRHKTTIFTDAKESSTVFELKRIVEGILKRPPDEQRLYKDDQLLDDGKTLGECGFTSQTARPQAPATV
GLAFDTFEALCIEPFSSPPEMYVKLISSDGHEFIVKREHALTSGTIKAMLSGPNEVNFREIPSHVLSKVCMYFTY
KVRYTNSSTEIPEFPIAPEIALELLMAANFLDRVVPDQRSKFENEEFFRKLSRECEIKYTGFRDRPHEERQARFQ
NACRDGRSEIAFVATGTNLSLQFFPASWREYVDLEREAGKVYLKAPMILNGVCVIWKGWIDLQRLDGMGCLEFDE
ERAQQEDALAQQAFEEARRRTREFEDRDMENRWQVMIVWQVDRMRINTWKRLVKHHMYISRKAKDWFYRHHYEST
NPKISSEVHIPLGDAKLVITTYWGLHTGERDWHLGQGVSIEWRKKRYSTQVDPDLADQLIHLHYFDCFSESAIRN
TILGRIVSPRCEYQAGHNKVGSLQYLALAALIKPKQIKPPLPSVRKLTED
PDB sequence matching range provided in alignment:
MDVFLMIRRHKTTIFTDAKESSTVFELKRIVEGILKRPPDEQRLYKDDQLLDDGKTLGECGFTSQTARPQAPATV
GLAFDTFEALCIEPFSSPPERVVPDQRSKFENEEFFRKLSRECEIKYTGFRDRPHEERQARFQNACRDGRSEIAF
VATGTNLSLQFFPASWREYVDLEREAGKVYLKAPMILNGVCVIWKGWIDLQRLDGMGCLEFDEERAQQEDALAQQ
AFEEARRRTREFEDRDMENRWQVMIVWQVDRMRINTWKRLVKHHMYISRKAKDWFYRHHYESTNPKISSEVHIPL
GDAKLVITTYWGLHTGERDWHLGQGVSIEWRKKRYSTQVDPDLADQLIHLHYFDCFSESAIRNTILGRIVSPRCE
YQAGHNKVGSLQYLALAALIKPKQIKPPLPSVRKLTED
No output models from Modeller; see log for Modeller text output.
> help help:user
> select clear
> help help:user
> ui tool show "Modeller Comparative"
> modeller comparative FL-CBFb_4n9f-CBFb.aln:1 FL-ELOB_4n9f-ELOB.aln:1 FL-
> ELOC_4n9f-ELOC.aln:1 FL-Hxb2Vif_4n9f-NL43.aln:1 multichain true numModels 5
> fast false hetPreserve false hydrogens false waterPreserve false
Web Service: Modeller9v8 is a Python wrapper that calls Modeller (v9.18) for
protein structure modeling
Opal service URL:
http://webservices.rbvi.ucsf.edu/opal2/services/Modeller9v8Service
Opal job id: appModeller9v8Service16287559492801691508076
Opal status URL prefix:
http://webservices.rbvi.ucsf.edu/appModeller9v8Service16287559492801691508076
stdout.txt = standard output
stderr.txt = standard error
Modeller job ID appModeller9v8Service16287559492801691508076 finished
Modeller error output
Traceback (most recent call last):
File "/usr/local/opal-local/bin/modeller9v8.py", line 322, in <module>
main()
File "/usr/local/opal-local/bin/modeller9v8.py", line 24, in main
VersionMap[cf["version"]](cf)
File "/usr/local/opal-local/bin/modeller9v8.py", line 34, in v2_run
execfile(fn)
File "ModellerModelling.py", line 67, in <module>
a.make()
File "/usr/lib64/python2.7/site-packages/modeller/automodel/automodel.py",
line 133, in make
self.homcsr(exit_stage)
File "/usr/lib64/python2.7/site-packages/modeller/automodel/automodel.py",
line 595, in homcsr
aln = self.read_alignment()
File "/usr/lib64/python2.7/site-packages/modeller/automodel/automodel.py",
line 556, in read_alignment
aln.append(file=self.alnfile, align_codes=codes)
File "/usr/lib64/python2.7/site-packages/modeller/alignment.py", line 80, in
append
allow_alternates)
_modeller.SequenceMismatchError: get_ran_648E> Alignment sequence does not
match that in PDB file: 1 ./4n9f.pdb1_1.pdb (You didn't specify the starting
and ending residue numbers and chain IDs in the alignment, so Modeller tried
to guess these from the PDB file.) Suggestion: put in the residue numbers and
chain IDs (see the manual) and run again for more detailed diagnostics. You
could also try running with allow_alternates=True to accept alternate one-
letter code matches (e.g. B to N, Z to Q).
Modeller run output
MODELLER 9.22, 2019/06/19, r11413
PROTEIN STRUCTURE MODELLING BY SATISFACTION OF SPATIAL RESTRAINTS
Copyright(c) 1989-2019 Andrej Sali
All Rights Reserved
Written by A. Sali
with help from
B. Webb, M.S. Madhusudhan, M-Y. Shen, G.Q. Dong,
M.A. Marti-Renom, N. Eswar, F. Alber, M. Topf, B. Oliva,
A. Fiser, R. Sanchez, B. Yerkovich, A. Badretdinov,
F. Melo, J.P. Overington, E. Feyfant
University of California, San Francisco, USA
Rockefeller University, New York, USA
Harvard University, Cambridge, USA
Imperial Cancer Research Fund, London, UK
Birkbeck College, University of London, London, UK
Kind, OS, HostName, Kernel, Processor: 4, Linux franklin.cgl.ucsf.edu
3.10.0-1160.36.2.el7.x86_64 x86_64
Date and time of compilation : 2019/06/19 14:00:54
MODELLER executable type : x86_64-intel8
Job starting time (YY/MM/DD HH:MM:SS): 2021/08/12 01:12:29
openf___224_> Open $(LIB)/restyp.lib
openf___224_> Open ${MODINSTALL9v22}/modlib/resgrp.lib
rdresgr_266_> Number of residue groups: 2
openf___224_> Open ${MODINSTALL9v22}/modlib/sstruc.lib
Dynamically allocated memory at amaxlibraries [B,KiB,MiB]: 191566 187.076
0.183
Dynamically allocated memory at amaxlibraries [B,KiB,MiB]: 192094 187.592
0.183
openf___224_> Open ${MODINSTALL9v22}/modlib/resdih.lib
Dynamically allocated memory at amaxlibraries [B,KiB,MiB]: 240694 235.053
0.230
rdrdih__263_> Number of dihedral angle types : 9
Maximal number of dihedral angle optima: 3
Dihedral angle names : Alph Phi Psi Omeg chi1 chi2 chi3 chi4 chi5
openf___224_> Open ${MODINSTALL9v22}/modlib/radii.lib
Dynamically allocated memory at amaxlibraries [B,KiB,MiB]: 253994 248.041
0.242
openf___224_> Open ${MODINSTALL9v22}/modlib/radii14.lib
openf___224_> Open ${MODINSTALL9v22}/modlib/af_mnchdef.lib
rdwilmo_274_> Mainchain residue conformation classes: APBLE
openf___224_> Open ${MODINSTALL9v22}/modlib/mnch.lib
rdclass_257_> Number of classes: 5
openf___224_> Open ${MODINSTALL9v22}/modlib/mnch1.lib
openf___224_> Open ${MODINSTALL9v22}/modlib/mnch2.lib
openf___224_> Open ${MODINSTALL9v22}/modlib/mnch3.lib
openf___224_> Open ${MODINSTALL9v22}/modlib/xs4.mat
rdrrwgh_268_> Number of residue types: 21
openf___224_> Open alignment.ali
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 269801 263.478
0.257
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 284893 278.216
0.272
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 300293 293.255
0.286
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 331093 323.333
0.316
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 392693 383.489
0.375
Dynamically allocated memory at amaxalignment [B,KiB,MiB]: 515893 503.802
0.492
openf___224_> Open ./4n9f.pdb1_1.pdb
get_ran_648E> Alignment sequence does not match that in PDB file: 1
./4n9f.pdb1_1.pdb
(You didn't specify the starting and ending residue numbers and
chain IDs in the alignment, so Modeller tried to guess these from
the PDB file.)
Suggestion: put in the residue numbers and chain IDs (see the
manual) and run again for more detailed diagnostics.
You could also try running with allow_alternates=True to accept
alternate one-letter code matches (e.g. B to N, Z to Q).
Alignment sequence:
MDVFLMIRRHKTTIFTDAKESSTVFELKRIVEGILKRPPDEQRLYKDDQLLDDGKTLGECGFTSQTARPQAPATV
GLAFDTFEALCIEPFSSPPEMYVKLISSDGHEFIVKREHALTSGTIKAMLSGPNEVNFREIPSHVLSKVCMYFTY
KVRYTNSSTEIPEFPIAPEIALELLMAANFLDRVVPDQRSKFENEEFFRKLSRECEIKYTGFRDRPHEERQARFQ
NACRDGRSEIAFVATGTNLSLQFFPASWREYVDLEREAGKVYLKAPMILNGVCVIWKGWIDLQRLDGMGCLEFDE
ERAQQEDALAQQAFEEARRRTREFEDRDMENRWQVMIVWQVDRMRINTWKRLVKHHMYISRKAKDWFYRHHYEST
NPKISSEVHIPLGDAKLVITTYWGLHTGERDWHLGQGVSIEWRKKRYSTQVDPDLADQLIHLHYFDCFSESAIRN
TILGRIVSPRCEYQAGHNKVGSLQYLALAALIKPKQIKPPLPSVRKLTED
PDB sequence matching range provided in alignment:
MDVFLMIRRHKTTIFTDAKESSTVFELKRIVEGILKRPPDEQRLYKDDQLLDDGKTLGECGFTSQTARPQAPATV
GLAFDTFEALCIEPFSSPPERVVPDQRSKFENEEFFRKLSRECEIKYTGFRDRPHEERQARFQNACRDGRSEIAF
VATGTNLSLQFFPASWREYVDLEREAGKVYLKAPMILNGVCVIWKGWIDLQRLDGMGCLEFDEERAQQEDALAQQ
AFEEARRRTREFEDRDMENRWQVMIVWQVDRMRINTWKRLVKHHMYISRKAKDWFYRHHYESTNPKISSEVHIPL
GDAKLVITTYWGLHTGERDWHLGQGVSIEWRKKRYSTQVDPDLADQLIHLHYFDCFSESAIRNTILGRIVSPRCE
YQAGHNKVGSLQYLALAALIKPKQIKPPLPSVRKLTED
No output models from Modeller; see log for Modeller text output.
OpenGL version: 3.3.0 NVIDIA 461.92
OpenGL renderer: GeForce RTX 2060 SUPER/PCIe/SSE2
OpenGL vendor: NVIDIA Corporation
Manufacturer: System manufacturer
Model: System Product Name
OS: Microsoft Windows 10 Home (Build 19042)
Memory: 34,261,569,536
MaxProcessMemory: 137,438,953,344
CPU: 16 Intel(R) Core(TM) i9-9900K CPU @ 3.60GHz
OSLanguage: en-US
Locale: ('zh_TW', 'cp950')
PyQt5 5.15.2, Qt 5.15.2
Installed Packages:
alabaster: 0.7.12
appdirs: 1.4.4
Babel: 2.9.1
backcall: 0.2.0
blockdiag: 2.0.1
certifi: 2020.12.5
cftime: 1.5.0
chardet: 3.0.4
ChimeraX-AddCharge: 1.0.1
ChimeraX-AddH: 2.1.6
ChimeraX-AlignmentAlgorithms: 2.0
ChimeraX-AlignmentHdrs: 3.2
ChimeraX-AlignmentMatrices: 2.0
ChimeraX-Alignments: 2.1
ChimeraX-AmberInfo: 1.0
ChimeraX-Arrays: 1.0
ChimeraX-Atomic: 1.13.2
ChimeraX-AtomicLibrary: 3.1.3
ChimeraX-AtomSearch: 2.0
ChimeraX-AtomSearchLibrary: 1.0
ChimeraX-AxesPlanes: 2.0
ChimeraX-BasicActions: 1.1
ChimeraX-BILD: 1.0
ChimeraX-BlastProtein: 1.1
ChimeraX-BondRot: 2.0
ChimeraX-BugReporter: 1.0
ChimeraX-BuildStructure: 2.5.2
ChimeraX-Bumps: 1.0
ChimeraX-BundleBuilder: 1.1
ChimeraX-ButtonPanel: 1.0
ChimeraX-CageBuilder: 1.0
ChimeraX-CellPack: 1.0
ChimeraX-Centroids: 1.1
ChimeraX-ChemGroup: 2.0
ChimeraX-Clashes: 2.1
ChimeraX-ColorActions: 1.0
ChimeraX-ColorGlobe: 1.0
ChimeraX-ColorKey: 1.2.1
ChimeraX-CommandLine: 1.1.4
ChimeraX-ConnectStructure: 2.0
ChimeraX-Contacts: 1.0
ChimeraX-Core: 1.2.5
ChimeraX-CoreFormats: 1.0
ChimeraX-coulombic: 1.1.1
ChimeraX-Crosslinks: 1.0
ChimeraX-Crystal: 1.0
ChimeraX-CrystalContacts: 1.0
ChimeraX-DataFormats: 1.1
ChimeraX-Dicom: 1.0
ChimeraX-DistMonitor: 1.1.3
ChimeraX-DistUI: 1.0
ChimeraX-Dssp: 2.0
ChimeraX-EMDB-SFF: 1.0
ChimeraX-ExperimentalCommands: 1.0
ChimeraX-FileHistory: 1.0
ChimeraX-FunctionKey: 1.0
ChimeraX-Geometry: 1.1
ChimeraX-gltf: 1.0
ChimeraX-Graphics: 1.0
ChimeraX-Hbonds: 2.1
ChimeraX-Help: 1.1
ChimeraX-HKCage: 1.3
ChimeraX-IHM: 1.0
ChimeraX-ImageFormats: 1.1
ChimeraX-IMOD: 1.0
ChimeraX-IO: 1.0.1
ChimeraX-Label: 1.0
ChimeraX-ListInfo: 1.1.1
ChimeraX-Log: 1.1.2
ChimeraX-LookingGlass: 1.1
ChimeraX-Maestro: 1.8.1
ChimeraX-Map: 1.0.2
ChimeraX-MapData: 2.0
ChimeraX-MapEraser: 1.0
ChimeraX-MapFilter: 2.0
ChimeraX-MapFit: 2.0
ChimeraX-MapSeries: 2.0
ChimeraX-Markers: 1.0
ChimeraX-Mask: 1.0
ChimeraX-MatchMaker: 1.2.1
ChimeraX-MDcrds: 2.2
ChimeraX-MedicalToolbar: 1.0.1
ChimeraX-Meeting: 1.0
ChimeraX-MLP: 1.1
ChimeraX-mmCIF: 2.3
ChimeraX-MMTF: 2.1
ChimeraX-Modeller: 1.0.1
ChimeraX-ModelPanel: 1.0.1
ChimeraX-ModelSeries: 1.0
ChimeraX-Mol2: 2.0
ChimeraX-Morph: 1.0
ChimeraX-MouseModes: 1.1
ChimeraX-Movie: 1.0
ChimeraX-Neuron: 1.0
ChimeraX-Nucleotides: 2.0.1
ChimeraX-OpenCommand: 1.5
ChimeraX-PDB: 2.4.1
ChimeraX-PDBBio: 1.0
ChimeraX-PDBLibrary: 1.0.1
ChimeraX-PDBMatrices: 1.0
ChimeraX-PickBlobs: 1.0
ChimeraX-Positions: 1.0
ChimeraX-PresetMgr: 1.0.1
ChimeraX-PubChem: 2.0.1
ChimeraX-ReadPbonds: 1.0
ChimeraX-Registration: 1.1
ChimeraX-RemoteControl: 1.0
ChimeraX-ResidueFit: 1.0
ChimeraX-RestServer: 1.1
ChimeraX-RNALayout: 1.0
ChimeraX-RotamerLibMgr: 2.0
ChimeraX-RotamerLibsDunbrack: 2.0
ChimeraX-RotamerLibsDynameomics: 2.0
ChimeraX-RotamerLibsRichardson: 2.0
ChimeraX-SaveCommand: 1.4
ChimeraX-SchemeMgr: 1.0
ChimeraX-SDF: 2.0
ChimeraX-Segger: 1.0
ChimeraX-Segment: 1.0
ChimeraX-SeqView: 2.3
ChimeraX-Shape: 1.0.1
ChimeraX-Shell: 1.0
ChimeraX-Shortcuts: 1.0
ChimeraX-ShowAttr: 1.0
ChimeraX-ShowSequences: 1.0
ChimeraX-SideView: 1.0
ChimeraX-Smiles: 2.0.1
ChimeraX-SmoothLines: 1.0
ChimeraX-SpaceNavigator: 1.0
ChimeraX-StdCommands: 1.3.1
ChimeraX-STL: 1.0
ChimeraX-Storm: 1.0
ChimeraX-Struts: 1.0
ChimeraX-Surface: 1.0
ChimeraX-SwapAA: 2.0
ChimeraX-SwapRes: 2.1
ChimeraX-TapeMeasure: 1.0
ChimeraX-Test: 1.0
ChimeraX-Toolbar: 1.0.1
ChimeraX-ToolshedUtils: 1.2
ChimeraX-Tug: 1.0
ChimeraX-UI: 1.7.6
ChimeraX-uniprot: 2.1
ChimeraX-UnitCell: 1.0
ChimeraX-ViewDockX: 1.0
ChimeraX-Vive: 1.1
ChimeraX-VolumeMenu: 1.0
ChimeraX-VTK: 1.0
ChimeraX-WavefrontOBJ: 1.0
ChimeraX-WebCam: 1.0
ChimeraX-WebServices: 1.0
ChimeraX-Zone: 1.0
colorama: 0.4.3
comtypes: 1.1.7
cxservices: 1.0
cycler: 0.10.0
Cython: 0.29.21
decorator: 5.0.9
distlib: 0.3.1
docutils: 0.16
filelock: 3.0.12
funcparserlib: 0.3.6
grako: 3.16.5
h5py: 2.10.0
html2text: 2020.1.16
idna: 2.10
ihm: 0.17
imagecodecs: 2020.5.30
imagesize: 1.2.0
ipykernel: 5.3.4
ipython: 7.18.1
ipython-genutils: 0.2.0
jedi: 0.17.2
Jinja2: 2.11.2
jupyter-client: 6.1.7
jupyter-core: 4.7.1
kiwisolver: 1.3.1
line-profiler: 2.1.2
lxml: 4.6.2
lz4: 3.1.0
MarkupSafe: 2.0.1
matplotlib: 3.3.2
msgpack: 1.0.0
netCDF4: 1.5.4
networkx: 2.5
numexpr: 2.7.3
numpy: 1.19.2
numpydoc: 1.1.0
openvr: 1.14.1501
packaging: 20.9
ParmEd: 3.2.0
parso: 0.7.1
pickleshare: 0.7.5
Pillow: 7.2.0
pip: 21.0.1
pkginfo: 1.5.0.1
prompt-toolkit: 3.0.18
psutil: 5.7.2
pycollada: 0.7.1
pydicom: 2.0.0
Pygments: 2.7.1
PyOpenGL: 3.1.5
PyOpenGL-accelerate: 3.1.5
pyparsing: 2.4.7
PyQt5-commercial: 5.15.2
PyQt5-sip: 12.8.1
PyQtWebEngine-commercial: 5.15.2
python-dateutil: 2.8.1
pytz: 2021.1
pywin32: 228
pyzmq: 22.0.3
qtconsole: 4.7.7
QtPy: 1.9.0
RandomWords: 0.3.0
requests: 2.24.0
scipy: 1.5.2
setuptools: 50.3.2
sfftk-rw: 0.6.7.dev1
six: 1.15.0
snowballstemmer: 2.1.0
sortedcontainers: 2.2.2
Sphinx: 3.2.1
sphinxcontrib-applehelp: 1.0.2
sphinxcontrib-blockdiag: 2.0.0
sphinxcontrib-devhelp: 1.0.2
sphinxcontrib-htmlhelp: 2.0.0
sphinxcontrib-jsmath: 1.0.1
sphinxcontrib-qthelp: 1.0.3
sphinxcontrib-serializinghtml: 1.1.5
suds-jurko: 0.6
tables: 3.6.1
tifffile: 2020.9.3
tinyarray: 1.2.3
tornado: 6.1
traitlets: 5.0.5
urllib3: 1.25.11
wcwidth: 0.2.5
webcolors: 1.11.1
wheel: 0.36.0
wheel-filename: 1.3.0
WMI: 1.5.1
File attachment: Modeller Comparative Error.png
{kind=link}
{kind=link}
Attachments (1)
Change History (8)
by , 5 years ago
| Attachment: | Modeller Comparative Error.png added |
|---|
comment:1 by , 5 years ago
| Component: | Unassigned → Sequence |
|---|---|
| Owner: | set to |
| Platform: | → all |
| Project: | → ChimeraX |
| Status: | new → accepted |
| Summary: | ChimeraX bug report submission → Modeller sequence mismatch failure |
comment:2 by , 5 years ago
I am in communication with the Modeller group, and it's still a little unclear at this point whether this is a Modeller problem or a ChimeraX problem. Until this is fixed, I believe you have to delete parts of the complex that are not being used as templates. I don't know if the resulting models will be satisfactory or not -- but I believe the job will run.
comment:3 by , 5 years ago
Thank you for your help, Eric. I would like to build a homology model for a tetrameric complex. I could build homology models using two proteins as templates, but the other two proteins always have issues. I'm looking forward to hearing that if the issue can be resolved in the future. Thank you.
Yen-Li
________________________________
From: ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu>
Sent: Friday, August 13, 2021 6:12 PM
Cc: PETTERSEN, ERIC <pett@cgl.ucsf.edu>; Li, Yen-Li <Yen-Li.Li@ucsf.edu>
Subject: Re: [ChimeraX] #5045: Modeller sequence mismatch failure
#5045: Modeller sequence mismatch failure
----------------------------------+----------------------------
Reporter: yen-li.li@… | Owner: Eric Pettersen
Type: defect | Status: accepted
Priority: normal | Milestone:
Component: Sequence | Version:
Resolution: | Keywords:
Blocked By: | Blocking:
Notify when closed: | Platform: all
Project: ChimeraX |
----------------------------------+----------------------------
Comment (by Eric Pettersen):
I am in communication with the Modeller group, and it's still a little
unclear at this point whether this is a Modeller problem or a ChimeraX
problem. Until this is fixed, I believe you have to delete parts of the
complex that are not being used as templates. I don't know if the
resulting models will be satisfactory or not -- but I believe the job will
run.
--
Ticket URL: <https://www.rbvi.ucsf.edu/trac/ChimeraX/ticket/5045#comment:2>
ChimeraX <https://www.rbvi.ucsf.edu/chimerax/>
ChimeraX Issue Tracker
follow-up: 3 comment:4 by , 5 years ago
| Resolution: | → fixed |
|---|---|
| Status: | accepted → closed |
Tomorrow's daily build should work without the delete-chains hocus pocus I mentioned before.
comment:5 by , 5 years ago
Hi Eric,
Thank you for the update. Is it possible to renumber the residue number of chains in Chimera X? After building homology model for two proteins (chain A: 182 residues; chain B: 192 residues) with modeller comparative, these two proteins were combined into a single model file and chain B was renumbered from 183-374. I would like to make chain B residues starting from 1 to 192.
Thank you
Yen-Li
________________________________
From: ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu>
Sent: Monday, August 16, 2021 3:27 PM
Cc: PETTERSEN, ERIC <pett@cgl.ucsf.edu>; Li, Yen-Li <Yen-Li.Li@ucsf.edu>
Subject: Re: [ChimeraX] #5045: Modeller sequence mismatch failure
#5045: Modeller sequence mismatch failure
----------------------------------+----------------------------
Reporter: yen-li.li@… | Owner: Eric Pettersen
Type: defect | Status: closed
Priority: normal | Milestone:
Component: Sequence | Version:
Resolution: fixed | Keywords:
Blocked By: | Blocking:
Notify when closed: | Platform: all
Project: ChimeraX |
----------------------------------+----------------------------
Changes (by Eric Pettersen):
* status: accepted => closed
* resolution: => fixed
Comment:
Tomorrow's daily build should work without the delete-chains hocus pocus I
mentioned before.
--
Ticket URL: <https://www.rbvi.ucsf.edu/trac/ChimeraX/ticket/5045#comment:4>
ChimeraX <https://www.rbvi.ucsf.edu/chimerax/>
ChimeraX Issue Tracker
follow-up: 5 comment:6 by , 5 years ago
There is no command for renumbering residues in ChimeraX yet. It is really easy to do in ChimeraX's Python shell if you know Python. If you do know Python, let me know and I'll send instructions.
--Eric
comment:7 by , 5 years ago
Hi Eric,
Thank you for your email. I used chimera to renumber the residue and then imported the file to chimera X for subsequent work.
Best,
Yen-Li
________________________________
From: ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu>
Sent: Wednesday, August 25, 2021 11:20 AM
Cc: PETTERSEN, ERIC <pett@cgl.ucsf.edu>; Li, Yen-Li <Yen-Li.Li@ucsf.edu>
Subject: Re: [ChimeraX] #5045: Modeller sequence mismatch failure
#5045: Modeller sequence mismatch failure
----------------------------------+----------------------------
Reporter: yen-li.li@… | Owner: Eric Pettersen
Type: defect | Status: closed
Priority: normal | Milestone:
Component: Sequence | Version:
Resolution: fixed | Keywords:
Blocked By: | Blocking:
Notify when closed: | Platform: all
Project: ChimeraX |
----------------------------------+----------------------------
Comment (by Eric Pettersen):
There is no command for renumbering residues in ChimeraX yet. It is
really easy to do in ChimeraX's Python shell if you know Python. If you
do know Python, let me know and I'll send instructions.
--Eric
--
Ticket URL: <https://www.rbvi.ucsf.edu/trac/ChimeraX/ticket/5045#comment:6>
ChimeraX <https://www.rbvi.ucsf.edu/chimerax/>
ChimeraX Issue Tracker
Note:
See TracTickets
for help on using tickets.
Added by email2trac