Opened 18 months ago
Closed 18 months ago
#16805 closed defect (can't reproduce)
Selection intersection problem
| Reported by: | Owned by: | Tom Goddard | |
|---|---|---|---|
| Priority: | normal | Milestone: | |
| Component: | Structure Analysis | Version: | |
| Keywords: | Cc: | Eric Pettersen | |
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
The following bug report has been submitted:
Platform: macOS-14.7.3-arm64-arm-64bit
ChimeraX Version: 1.9 (2024-12-11 19:11:19 UTC)
Description
The intersect selection does not seem to be working.
For example, I want to colour the sidechains of residues in one chain that contact another chain:
1. select (replace) contacts between 2 chains : selects several residues in both chains. OK
2. select (substract) 1 chain : only contact residues in 1 chain are shown, including sidechains. OK
3. select (intersect) sidechain only: removes all selection! ctrl-Z does not restore selection :(
There are workaround, but still it would be really helpful to have the intersect selection work.
Log:
UCSF ChimeraX version: 1.9 (2024-12-11)
© 2016-2024 Regents of the University of California. All rights reserved.
> open
> /Users/iphan/Documents/ssgcid/OQ8b_HIP_42_neuraminidase/documents/neuraminidase_svtd.cxs
Log from Wed May 1 10:43:48 2024UCSF ChimeraX version: 1.7.1 (2024-01-23)
© 2016-2023 Regents of the University of California. All rights reserved.
> open "/Users/iphan/Documents/ssgcid/Flu
> project/neuraminidase/neuraminidase.cxs" format session
Log from Wed Dec 13 18:08:18 2023UCSF ChimeraX version: 1.6.1 (2023-05-09)
© 2016-2023 Regents of the University of California. All rights reserved.
> open "/Users/iphan/Documents/ssgcid/Flu
> project/neuraminidase/neuraminidase.cxs"
Log from Fri Dec 1 10:04:32 2023UCSF ChimeraX version: 1.6.1 (2023-05-09)
© 2016-2023 Regents of the University of California. All rights reserved.
> open "/Users/iphan/Documents/ssgcid/Flu
> project/neuraminidase/neuraminidase.cxs" format session
Log from Wed Nov 29 16:58:50 2023UCSF ChimeraX version: 1.6.1 (2023-05-09)
© 2016-2023 Regents of the University of California. All rights reserved.
How to cite UCSF ChimeraX
> open
> /Users/iphan/Documents/BaseJ/PAF1C/sequences/LEO1_PAF1_colabfold_batch/LEO1_Q8IJJ3_unrelaxed_alphafold2_multimer_v3_model_1_seed_000.pdb
Chain information for
LEO1_Q8IJJ3_unrelaxed_alphafold2_multimer_v3_model_1_seed_000.pdb #1
---
Chain | Description
A | No description available
B | No description available
> select add #1
6436 atoms, 6566 bonds, 760 residues, 1 model selected
> color sel bychain
> select clear
[Repeated 1 time(s)]
> select add #1
6436 atoms, 6566 bonds, 760 residues, 1 model selected
> color bfactor sel
6436 atoms, 760 residues, atom bfactor range 14.5 to 95.4
> select clear
> color bychain
> close
> open 6hg0
Summary of feedback from opening 6hg0 fetched from pdb
---
warnings | Unable to fetch template for 'BMA': might have incorrect bonds
Unable to fetch template for 'MAN': might have incorrect bonds
Unable to fetch template for 'PO4': might have incorrect bonds
Unable to fetch template for 'SIA': might have incorrect bonds
Unable to fetch template for 'K': might have incorrect bonds
note | Fetching compressed mmCIF 6hg0 from http://files.rcsb.org/download/6hg0.cif
6hg0 title:
Influenza A Virus N9 Neuraminidase complex with NANA (Tern/Australia). [more
info...]
Chain information for 6hg0 #1
---
Chain | Description | UniProt
A | Neuraminidase | NRAM_I75A5 83-470
Non-standard residues in 6hg0 #1
---
BMA — beta-D-mannopyranose
CA — calcium ion
GOL — glycerol (glycerin; propane-1,2,3-triol)
K — potassium ion
MAN — alpha-D-mannopyranose
NAG — 2-acetamido-2-deoxy-beta-D-glucopyranose
PO4 — phosphate ion
SIA — N-acetyl-alpha-neuraminic acid
6hg0 mmCIF Assemblies
---
1| author_and_software_defined_assembly
> select #1:119,120,135,152,153,181,227,228,229,279,294,351,372,406
130 atoms, 121 bonds, 14 residues, 1 model selected
> ui tool show "Show Sequence Viewer"
> sequence chain /A
Alignment identifier is 1/A
> select add #1
4040 atoms, 3411 bonds, 14 pseudobonds, 1139 residues, 2 models selected
> mlp sel
Map values for surface "6hg0_A SES surface": minimum -29.21, mean -5.382,
maximum 24.5
To also show corresponding color key, enter the above mlp command and add key
true
> select #1:119,120,135,152,153,181,227,228,229,279,294,351,372,406
130 atoms, 121 bonds, 14 residues, 1 model selected
> color (#!1 & sel) red
> undo
> select intersect sideonly
61 bonds, 1 model selected
> hide #1.2 models
> color sel red
> undo
> select #1:119,120,135,152,153,181,227,228,229,279,294,351,372,406
130 atoms, 121 bonds, 14 residues, 1 model selected
> select intersect sideonly
61 bonds, 1 model selected
> color sel red
[Repeated 1 time(s)]
> select up
74 atoms, 61 bonds, 14 residues, 1 model selected
> select down
61 bonds, 2 models selected
> select up
74 atoms, 61 bonds, 14 residues, 1 model selected
> color (#!1 & sel) red
> show #1.2 models
> select /A:467@OE2
1 atom, 1 residue, 1 model selected
> select up
9 atoms, 8 bonds, 1 residue, 2 models selected
> select up
58 atoms, 59 bonds, 6 residues, 2 models selected
> select up
3067 atoms, 3159 bonds, 388 residues, 2 models selected
> select up
3868 atoms, 3225 bonds, 1125 residues, 2 models selected
> select up
4040 atoms, 3411 bonds, 1139 residues, 2 models selected
> select up
4040 atoms, 3411 bonds, 14 pseudobonds, 1139 residues, 3 models selected
> select up
4040 atoms, 3411 bonds, 14 pseudobonds, 1139 residues, 3 models selected
> select up
4040 atoms, 3411 bonds, 14 pseudobonds, 1139 residues, 3 models selected
> select up
4040 atoms, 3411 bonds, 14 pseudobonds, 1139 residues, 3 models selected
> hide #1.2 models
> show #1.2 models
> hide #1.2 models
> show #1.2 models
> select /A:224@CD1
1 atom, 1 residue, 1 model selected
> select add /A:151@CE1
2 atoms, 2 residues, 2 models selected
> select up
18 atoms, 17 bonds, 2 residues, 2 models selected
> label (#!1 & sel) text "{0.label_one_letter_code}
> {0.number}{0.insertion_code}"
> select add /A:153@NH1
19 atoms, 17 bonds, 3 residues, 2 models selected
> select up
29 atoms, 27 bonds, 3 residues, 2 models selected
> label (#!1 & sel) text "{0.label_one_letter_code}
> {0.number}{0.insertion_code}"
> undo
[Repeated 8 time(s)]
> redo
> undo
> redo
[Repeated 7 time(s)]
> ~label (#!1 & sel) residues
> select /A:348@ND2
1 atom, 1 residue, 1 model selected
> select add /A:372@CD
2 atoms, 2 residues, 2 models selected
> select add /A:434@NZ
3 atoms, 3 residues, 2 models selected
> select up
28 atoms, 25 bonds, 3 residues, 2 models selected
> label (#!1 & sel) text "{0.label_one_letter_code}
> {0.number}{0.insertion_code}"
> ui mousemode right "move label"
> select /A:328@CD
1 atom, 1 residue, 1 model selected
> select /A:328@CG
1 atom, 1 residue, 1 model selected
> lighting full
> select /A:328@CB
1 atom, 1 residue, 1 model selected
> select /A:328@O
1 atom, 1 residue, 1 model selected
> select /A:328@CD
1 atom, 1 residue, 1 model selected
> select clear
> save "/Users/iphan/Documents/ssgcid/Flu
> project/neuraminidase/neuraminidase.cxs"
——— End of log from Wed Nov 29 16:58:50 2023 ———
opened ChimeraX session
> hide #1.2 models
> show #1.2 models
> select /A:372@NE
1 atom, 1 residue, 1 model selected
> select up
11 atoms, 10 bonds, 1 residue, 2 models selected
> select add /A:294@NH1
12 atoms, 10 bonds, 2 residues, 2 models selected
> select up
22 atoms, 20 bonds, 2 residues, 2 models selected
> select add /A:119@NH2
23 atoms, 20 bonds, 3 residues, 2 models selected
> select up
33 atoms, 30 bonds, 3 residues, 2 models selected
> select add /A:152@OD2
34 atoms, 30 bonds, 4 residues, 2 models selected
> select up
41 atoms, 37 bonds, 4 residues, 2 models selected
> select add /A:153@NH1
42 atoms, 37 bonds, 5 residues, 2 models selected
> select up
52 atoms, 48 bonds, 5 residues, 2 models selected
> hide #1.2 models
Drag select of 2 residues
> undo
> select up
54 atoms, 49 bonds, 6 residues, 2 models selected
> select up
64 atoms, 60 bonds, 6 residues, 2 models selected
> select add /A:279@CD
65 atoms, 60 bonds, 7 residues, 2 models selected
> select up
73 atoms, 68 bonds, 7 residues, 2 models selected
> show #1.2 models
> select add /A:229@CD
74 atoms, 68 bonds, 8 residues, 2 models selected
> select up
82 atoms, 76 bonds, 8 residues, 2 models selected
> select up
522 atoms, 526 bonds, 66 residues, 2 models selected
> select down
82 atoms, 76 bonds, 8 residues, 2 models selected
> select add /A:120@CG
83 atoms, 76 bonds, 9 residues, 2 models selected
> select up
91 atoms, 85 bonds, 9 residues, 2 models selected
> select up
522 atoms, 526 bonds, 66 residues, 2 models selected
> select down
91 atoms, 85 bonds, 9 residues, 2 models selected
> hide #1.2 models
> show sel atoms
> label (#!1 & sel) text "{0.label_one_letter_code}
> {0.number}{0.insertion_code}"
> show #1.2 models
> select #1:119,120,152,153,229,279,294,372,406
91 atoms, 85 bonds, 9 residues, 1 model selected
> select clear
> select #1:119,120,152,153,229,279,294,372,406
91 atoms, 85 bonds, 9 residues, 1 model selected
> open "/Users/iphan/Documents/ssgcid/Flu
> project/neuraminidase/flu_neuraminidase_100_hotspots_all_rfd.result/flu_neuraminidase_100_hotspots_all_rfd/best.pdb"
Chain information for best.pdb #2
---
Chain | Description
A | No description available
B | No description available
> hide #1.3 models
> hide #!1 models
> select add #2
4014 atoms, 4097 bonds, 497 residues, 3 models selected
> select subtract #1.2
3923 atoms, 4012 bonds, 488 residues, 2 models selected
> view sel
> cofr sel
> hide #2 models
> select subtract #2
Nothing selected
> show #!1 models
> hide #!1 models
> show #!1 models
> show #1.3 models
> select add #1
4040 atoms, 3411 bonds, 14 pseudobonds, 1139 residues, 3 models selected
> view sel
> cofr sel
> select #2/A
3066 atoms, 3149 bonds, 388 residues, 1 model selected
> hide #1.2 models
> select #1/A
3868 atoms, 3225 bonds, 9 pseudobonds, 1125 residues, 2 models selected
> select intersect
> ::name="ALA"::name="ARG"::name="ASN"::name="ASP"::name="CYS"::name="GLN"::name="GLU"::name="GLY"::name="HIS"::name="ILE"::name="LEU"::name="LYS"::name="MET"::name="PHE"::name="PRO"::name="SER"::name="THR"::name="TRP"::name="TYR"::name="VAL"
3159 bonds, 1 model selected
> select ~sel
7963 atoms, 7423 bonds, 14 pseudobonds, 1627 residues, 4 models selected
> select clear
> select intersect #1/A
Nothing selected
> select #1/A
3868 atoms, 3225 bonds, 9 pseudobonds, 1125 residues, 2 models selected
> select intersect main
3159 bonds, 1 model selected
> select up
3067 atoms, 3159 bonds, 388 residues, 1 model selected
> select up
3868 atoms, 3225 bonds, 1125 residues, 2 models selected
> select down
3067 atoms, 3159 bonds, 388 residues, 2 models selected
> save "/Users/iphan/Documents/ssgcid/Flu
> project/neuraminidase/6hg0_protein_monomer.pdb" models #1 selectedOnly true
> relModel #1
> hide #!1 models
> hide #1.1 models
> hide #1.3 models
> select add #1
4040 atoms, 3411 bonds, 14 pseudobonds, 1139 residues, 4 models selected
> select subtract #1
1 model selected
> open "/Users/iphan/Documents/ssgcid/Flu
> project/neuraminidase/6hg0_protein_monomer.pdb"
Summary of feedback from opening /Users/iphan/Documents/ssgcid/Flu
project/neuraminidase/6hg0_protein_monomer.pdb
---
warnings | Cannot find LINK/SSBOND residue NAG (1 )
Cannot find LINK/SSBOND residue NAG (1 )
Cannot find LINK/SSBOND residue NAG (1 )
Cannot find LINK/SSBOND residue NAG (1 )
Cannot find LINK/SSBOND residue NAG (2 )
23 messages similar to the above omitted
Chain information for 6hg0_protein_monomer.pdb #3
---
Chain | Description
A | No description available
> show #!1 models
> hide #!1 models
> show #!1 models
> hide #!3 models
> show #!3 models
> hide #!1 models
> select #1:119,120,152,153,229,279,294,372,406
91 atoms, 85 bonds, 9 residues, 1 model selected
> select #2:119,120,152,153,229,279,294,372,406
64 atoms, 60 bonds, 8 residues, 1 model selected
> select #3:119,120,152,153,229,279,294,372,406
91 atoms, 85 bonds, 9 residues, 1 model selected
> show sel atoms
> color sel red
> select add #3
3067 atoms, 3157 bonds, 2 pseudobonds, 388 residues, 2 models selected
> mlp sel
Map values for surface "6hg0_protein_monomer.pdb_A SES surface": minimum
-29.21, mean -5.382, maximum 24.5
To also show corresponding color key, enter the above mlp command and add key
true
> select #3:119,120,152,153,229,279,294,372,406
91 atoms, 85 bonds, 9 residues, 1 model selected
> color (#!3 & sel) red
> show #!1 models
> hide #!1 models
> show #!1 models
> hide #!1 models
> hide #!3 models
> show #!1 models
> show #1.2 models
> show #!3 models
> hide #1.2 models
> show #1.2 models
> hide #1.2 models
> show #1.2 models
> hide #1.2 models
> show #1.2 models
> hide #1.2 models
> show #1.2 models
> hide #1.2 models
> show #1.1 models
> hide #1.1 models
> hide #!3 models
> select add #3.1
91 atoms, 85 bonds, 2 pseudobonds, 9 residues, 3 models selected
> select subtract #3.1
91 atoms, 85 bonds, 9 residues, 2 models selected
> hide #3.1 models
> show #!3 models
> hide #3.2 models
> show #1.2 models
> show #3.2 models
> hide #3.2 models
> show #3.2 models
> hide #3.2 models
> show #3.2 models
> hide #3.2 models
> show #3.2 models
> hide #3.2 models
> show #3.2 models
> hide #3.2 models
> select #1/A:181@CB
1 atom, 1 residue, 1 model selected
> select up
6 atoms, 5 bonds, 1 residue, 2 models selected
> show #3.2 models
> hide #3.2 models
> show #3.2 models
> hide #3.2 models
> show #3.2 models
> hide #3.2 models
> show #3.2 models
> hide #3.2 models
> show #3.2 models
> hide #3.2 models
> hide #!3 models
> select #1/A:153@N
1 atom, 1 residue, 1 model selected
> select up
11 atoms, 10 bonds, 1 residue, 2 models selected
> select up
86 atoms, 87 bonds, 10 residues, 2 models selected
> select down
11 atoms, 10 bonds, 1 residue, 2 models selected
> hide #1.2 models
> show #1.2 models
> show #3.1 models
> hide #3.1 models
> show #3.2 models
> hide #3.2 models
> show #3.2 models
> hide #3.2 models
> show #3.2 models
> hide #3.2 models
> show #3.2 models
> hide #3.2 models
> show #3.2 models
> hide #3.2 models
> show #3.2 models
> hide #3.2 models
> show #3.2 models
> hide #3.2 models
> hide #!3 models
> select up
86 atoms, 87 bonds, 10 residues, 2 models selected
> select down
11 atoms, 10 bonds, 1 residue, 2 models selected
> select up
86 atoms, 87 bonds, 10 residues, 2 models selected
> select up
3067 atoms, 3159 bonds, 388 residues, 2 models selected
> select down
86 atoms, 87 bonds, 10 residues, 2 models selected
> select down
11 atoms, 10 bonds, 1 residue, 2 models selected
> color (#!1 & sel) red
> show #1.3 models
> show #3.2 models
> hide #3.2 models
> show #3.2 models
> hide #3.2 models
> show #3.2 models
> hide #1.2 models
> hide #!1 models
> select #3:119,120,152,153,229,279,294,372,406
91 atoms, 85 bonds, 9 residues, 1 model selected
> hide #3.2 models
> hide #!3 models
> show #1.2 models
> hide #!1 models
> show #!1 models
> hide #1.2 models
> hide #1.3 models
> hide #!1 models
> open
> /Users/iphan/Documents/BaseJ/PAF1C/sequences/LEO1_PAF1_colabfold_batch/LEO1_Q8I3K1_unrelaxed_rank_001_alphafold2_multimer_v3_model_3_seed_000.pdb
> /Users/iphan/Documents/BaseJ/PAF1C/sequences/LEO1_PAF1_colabfold_batch/LEO1_Q8I3K1_unrelaxed_rank_002_alphafold2_multimer_v3_model_4_seed_000.pdb
> /Users/iphan/Documents/BaseJ/PAF1C/sequences/LEO1_PAF1_colabfold_batch/LEO1_Q8I3K1_unrelaxed_rank_003_alphafold2_multimer_v3_model_1_seed_000.pdb
> /Users/iphan/Documents/BaseJ/PAF1C/sequences/LEO1_PAF1_colabfold_batch/LEO1_Q8I3K1_unrelaxed_rank_004_alphafold2_multimer_v3_model_5_seed_000.pdb
> /Users/iphan/Documents/BaseJ/PAF1C/sequences/LEO1_PAF1_colabfold_batch/LEO1_Q8I3K1_unrelaxed_rank_005_alphafold2_multimer_v3_model_2_seed_000.pdb
Chain information for
LEO1_Q8I3K1_unrelaxed_rank_001_alphafold2_multimer_v3_model_3_seed_000.pdb #4
---
Chain | Description
A | No description available
B | No description available
Chain information for
LEO1_Q8I3K1_unrelaxed_rank_002_alphafold2_multimer_v3_model_4_seed_000.pdb #5
---
Chain | Description
A | No description available
B | No description available
Chain information for
LEO1_Q8I3K1_unrelaxed_rank_003_alphafold2_multimer_v3_model_1_seed_000.pdb #6
---
Chain | Description
A | No description available
B | No description available
Chain information for
LEO1_Q8I3K1_unrelaxed_rank_004_alphafold2_multimer_v3_model_5_seed_000.pdb #7
---
Chain | Description
A | No description available
B | No description available
Chain information for
LEO1_Q8I3K1_unrelaxed_rank_005_alphafold2_multimer_v3_model_2_seed_000.pdb #8
---
Chain | Description
A | No description available
B | No description available
> hide #5 models
> hide #6 models
> hide #7 models
> hide #8 models
> show #5 models
> show #6 models
> show #7 models
> show #8 models
> select add #4
6503 atoms, 6614 bonds, 777 residues, 3 models selected
> select add #5
12915 atoms, 13143 bonds, 1545 residues, 4 models selected
> select subtract #3.2
12824 atoms, 13058 bonds, 1536 residues, 3 models selected
> select add #6
19236 atoms, 19587 bonds, 2304 residues, 3 models selected
> select add #7
25648 atoms, 26116 bonds, 3072 residues, 4 models selected
> select add #8
32060 atoms, 32645 bonds, 3840 residues, 5 models selected
> color sel bychain
> hide #8 models
> hide #7 models
> hide #6 models
> hide #5 models
> show #5 models
> hide #4 models
> show #6 models
> hide #5 models
> show #7 models
> hide #6 models
> lighting simple
> show #8 models
> hide #7 models
> close #4-8
> show #!3 models
> show #3.1 models
> show #3.2 models
> open "/Users/iphan/Documents/ssgcid/Flu
> project/neuraminidase/flu_neuraminidase_10_hotspots_9_rfd_beta.result/flu_neuraminidase_10_hotspots_9_rfd_beta/best.pdb"
Chain information for best.pdb #4
---
Chain | Description
A | No description available
B | No description available
> hide #!3 models
> hide #3.1 models
> hide #3.2 models
> select add #4
3136 atoms, 3219 bonds, 398 residues, 1 model selected
> cofr sel
> view sel
> color sel bychain
> select #4:119,120,152,153,229,279,294,372,406
64 atoms, 60 bonds, 8 residues, 1 model selected
> select #4:119,120,152,153,229,279,294,372,406
64 atoms, 60 bonds, 8 residues, 1 model selected
> select #4:119,120,152,153,229,279,294,372,406
64 atoms, 60 bonds, 8 residues, 1 model selected
> select #4:119,120,152,153,229,279,294,372,406
64 atoms, 60 bonds, 8 residues, 1 model selected
> show #!1 models
> select #1:119,120,152,153,229,279,294,372,406
91 atoms, 85 bonds, 9 residues, 1 model selected
> select #4:119,120,152,153,229,279,294,372,406
64 atoms, 60 bonds, 8 residues, 1 model selected
> open "/Users/iphan/Documents/ssgcid/Flu
> project/neuraminidase/6hg0_protein_monomer.pdb"
Summary of feedback from opening /Users/iphan/Documents/ssgcid/Flu
project/neuraminidase/6hg0_protein_monomer.pdb
---
warnings | Cannot find LINK/SSBOND residue NAG (1 )
Cannot find LINK/SSBOND residue NAG (1 )
Cannot find LINK/SSBOND residue NAG (1 )
Cannot find LINK/SSBOND residue NAG (1 )
Cannot find LINK/SSBOND residue NAG (2 )
23 messages similar to the above omitted
Chain information for 6hg0_protein_monomer.pdb #5
---
Chain | Description
A | No description available
> ui tool show "Show Sequence Viewer"
> sequence chain #5/A
Alignment identifier is 5/A
> ui tool show "Renumber Residues"
> renumber #4/A seqStart 83
388 residues renumbered
> select #4:119,120,152,153,229,279,294,372,406
91 atoms, 85 bonds, 9 residues, 1 model selected
> show sel atoms
> select #4/B:10
7 atoms, 7 bonds, 1 residue, 1 model selected
> select up
70 atoms, 70 bonds, 10 residues, 1 model selected
> show sel atoms
> color sel byhetero
> select #4:119,120,152,153,229,279,294,372,406
91 atoms, 85 bonds, 9 residues, 1 model selected
> color sel byhetero
> select clear
> select #4/B:5
5 atoms, 4 bonds, 1 residue, 1 model selected
> select up
70 atoms, 70 bonds, 10 residues, 1 model selected
> select up
3136 atoms, 3219 bonds, 398 residues, 1 model selected
> select down
70 atoms, 70 bonds, 10 residues, 1 model selected
> hide sel cartoons
> select #4:119,120,152,153,229,279,294,372,406
91 atoms, 85 bonds, 9 residues, 1 model selected
> select clear
> select #4/A:346
8 atoms, 7 bonds, 1 residue, 1 model selected
> select up
293 atoms, 301 bonds, 39 residues, 1 model selected
> select up
3066 atoms, 3149 bonds, 388 residues, 1 model selected
> mlp sel
Map values for surface "best.pdb_A SES surface": minimum -27.51, mean -5.14,
maximum 24.64
To also show corresponding color key, enter the above mlp command and add key
true
> select up
2 atoms, 1 bond, 1 residue, 1 model selected
> select up
7 atoms, 6 bonds, 1 residue, 1 model selected
> select up
70 atoms, 70 bonds, 10 residues, 1 model selected
> surface sel
> transparency (#!4 & sel) 40
> select #4/A:348@ND2
1 atom, 1 residue, 1 model selected
> select up
8 atoms, 7 bonds, 1 residue, 2 models selected
> select up
293 atoms, 301 bonds, 39 residues, 2 models selected
> select up
3066 atoms, 3149 bonds, 388 residues, 2 models selected
> select down
293 atoms, 301 bonds, 39 residues, 2 models selected
> select #4/A:346@O
1 atom, 1 residue, 1 model selected
> select up
8 atoms, 7 bonds, 1 residue, 2 models selected
> select up
293 atoms, 301 bonds, 39 residues, 2 models selected
> select up
3066 atoms, 3149 bonds, 388 residues, 2 models selected
> select down
293 atoms, 301 bonds, 39 residues, 2 models selected
> hide #!5 models
> select add #4
3136 atoms, 3219 bonds, 398 residues, 2 models selected
> hide #4.1 models
> show #4.1 models
> surface style #4 mesh
> undo
> redo
> surface style #4 solid
> select subtract #4.1
70 atoms, 70 bonds, 10 residues, 3 models selected
> select add #4.1
3136 atoms, 70 bonds, 398 residues, 2 models selected
> select subtract #4.2
3066 atoms, 388 residues, 3 models selected
> select add #4
3136 atoms, 3219 bonds, 398 residues, 2 models selected
> select subtract #4
2 models selected
> select add #4.1
3066 atoms, 388 residues, 1 model selected
> select subtract #4.1
1 model selected
> select add #4.2
70 atoms, 10 residues, 1 model selected
> select subtract #4.2
1 model selected
> select add #4.1
3066 atoms, 388 residues, 1 model selected
> select subtract #4.1
1 model selected
> hide #4.1 models
> select #4/A:405
4 atoms, 3 bonds, 1 residue, 1 model selected
> select up
67 atoms, 69 bonds, 8 residues, 2 models selected
> select up
3066 atoms, 3149 bonds, 388 residues, 2 models selected
> select up
3136 atoms, 3219 bonds, 398 residues, 2 models selected
> select down
3066 atoms, 3149 bonds, 388 residues, 3 models selected
> hide #4.2 models
> show #4.2 models
> show #4.1 models
> transparency (#!4 & sel) 40
> hide #!1 models
> select #4/A:328@CB
1 atom, 1 residue, 1 model selected
> ui tool show "Side View"
> select #4:119,120,152,153,229,279,294,372,406
91 atoms, 85 bonds, 9 residues, 1 model selected
> hide #!4.1 models
> select #4:153,406
23 atoms, 22 bonds, 2 residues, 1 model selected
> select #4:153,372,406
34 atoms, 32 bonds, 3 residues, 1 model selected
> save "/Users/iphan/Documents/ssgcid/Flu
> project/neuraminidase/neuraminidase.cxs"
——— End of log from Fri Dec 1 10:04:32 2023 ———
opened ChimeraX session
> lighting full
> select clear
> select #4/A:279@CD
1 atom, 1 residue, 1 model selected
> select add #4/A:229@CD
2 atoms, 2 residues, 2 models selected
> select add #4/A:406@OH
3 atoms, 1 bond, 3 residues, 2 models selected
> select add #4/A:372@CZ
4 atoms, 1 bond, 4 residues, 2 models selected
> select up
6 atoms, 1 bond, 5 residues, 2 models selected
> select up
8 atoms, 2 bonds, 6 residues, 2 models selected
> select up
63 atoms, 58 bonds, 6 residues, 2 models selected
> select up
65 atoms, 59 bonds, 7 residues, 2 models selected
> select up
72 atoms, 67 bonds, 7 residues, 2 models selected
> select up
74 atoms, 68 bonds, 8 residues, 2 models selected
> select up
83 atoms, 77 bonds, 8 residues, 2 models selected
> ui tool show "Side View"
> set bgColor white
> set bgColor black
> set bgColor white
> save /Users/iphan/Desktop/image1.png supersample 3
> show #2 models
> hide #!4 models
> select add #4
3136 atoms, 3219 bonds, 398 residues, 2 models selected
> show #!4 models
> ui tool show Matchmaker
> matchmaker #2 to #4
Parameters
---
Chain pairing | bb
Alignment algorithm | Needleman-Wunsch
Similarity matrix | BLOSUM-62
SS fraction | 0.3
Gap open (HH/SS/other) | 18/18/6
Gap extend | 1
SS matrix | | | H | S | O
---|---|---|---
H | 6 | -9 | -6
S | | 6 | -6
O | | | 4
Iteration cutoff | 2
Matchmaker best.pdb, chain A (#4) with best.pdb, chain A (#2), sequence
alignment score = 2075.4
RMSD between 388 pruned atom pairs is 0.092 angstroms; (across all 388 pairs:
0.092)
> hide #!4 models
> select #2/A:263
8 atoms, 7 bonds, 1 residue, 1 model selected
> select up
293 atoms, 301 bonds, 39 residues, 1 model selected
> select up
3066 atoms, 3149 bonds, 388 residues, 1 model selected
> select up
3923 atoms, 4012 bonds, 488 residues, 1 model selected
> select down
3066 atoms, 3149 bonds, 388 residues, 1 model selected
> color sel bychain
> select clear
> show #!4 models
> select add #4/A:229@CG
1 atom, 1 residue, 1 model selected
> select add #4/A:279@CG
2 atoms, 2 residues, 2 models selected
> select add #4/A:153@CZ
3 atoms, 3 residues, 2 models selected
> hide #!4 models
> select add #2/B:61
12 atoms, 10 bonds, 4 residues, 3 models selected
> select up
16 atoms, 10 bonds, 6 residues, 3 models selected
> select up
59 atoms, 54 bonds, 6 residues, 3 models selected
> select up
852 atoms, 860 bonds, 101 residues, 3 models selected
> select up
3923 atoms, 4012 bonds, 488 residues, 3 models selected
> surface (#2 & sel)
> transparency (#!2 & sel) 30
> select clear
> select #2/B:59@OE1
1 atom, 1 residue, 1 model selected
> select up
9 atoms, 8 bonds, 1 residue, 2 models selected
> select up
380 atoms, 383 bonds, 42 residues, 2 models selected
> select up
857 atoms, 863 bonds, 100 residues, 2 models selected
> transparency (#!2 & sel) 50
> select clear
> show #!4 models
> select add #4/A:135
8 atoms, 9 bonds, 1 residue, 1 model selected
> undo
> select add #4/A:120@CG
1 atom, 2 bonds, 1 residue, 1 model selected
> select add #4/A:294@CZ
2 atoms, 2 bonds, 2 residues, 2 models selected
> select up
10 atoms, 4 bonds, 6 residues, 2 models selected
> select up
63 atoms, 58 bonds, 6 residues, 2 models selected
> list selected
Unknown command: list selected
> list res selected
Unknown command: list res selected
> list
Unknown command: list
> info selection
atom id #4/A:120@N idatm_type Npl
atom id #4/A:120@CA idatm_type C3
atom id #4/A:120@C idatm_type C2
atom id #4/A:120@CB idatm_type C3
atom id #4/A:120@O idatm_type O2
atom id #4/A:120@CG idatm_type C3
atom id #4/A:120@CD idatm_type Cac
atom id #4/A:120@OE1 idatm_type O2-
atom id #4/A:120@OE2 idatm_type O2-
atom id #4/A:153@N idatm_type Npl
atom id #4/A:153@CA idatm_type C3
atom id #4/A:153@C idatm_type C2
atom id #4/A:153@CB idatm_type C3
atom id #4/A:153@O idatm_type O2
atom id #4/A:153@CG idatm_type C3
atom id #4/A:153@CD idatm_type C3
atom id #4/A:153@NE idatm_type Ng+
atom id #4/A:153@NH1 idatm_type Ng+
atom id #4/A:153@NH2 idatm_type Ng+
atom id #4/A:153@CZ idatm_type C2
atom id #4/A:229@N idatm_type Npl
atom id #4/A:229@CA idatm_type C3
atom id #4/A:229@C idatm_type C2
atom id #4/A:229@CB idatm_type C3
atom id #4/A:229@O idatm_type O2
atom id #4/A:229@CG idatm_type C3
atom id #4/A:229@CD idatm_type Cac
atom id #4/A:229@OE1 idatm_type O2-
atom id #4/A:229@OE2 idatm_type O2-
atom id #4/A:294@N idatm_type Npl
atom id #4/A:294@CA idatm_type C3
atom id #4/A:294@C idatm_type C2
atom id #4/A:294@CB idatm_type C3
atom id #4/A:294@O idatm_type O2
atom id #4/A:294@CG idatm_type C3
atom id #4/A:294@CD idatm_type C3
atom id #4/A:294@NE idatm_type Ng+
atom id #4/A:294@NH1 idatm_type Ng+
atom id #4/A:294@NH2 idatm_type Ng+
atom id #4/A:294@CZ idatm_type C2
atom id #4/A:372@N idatm_type Npl
atom id #4/A:372@CA idatm_type C3
atom id #4/A:372@C idatm_type C2
atom id #4/A:372@CB idatm_type C3
atom id #4/A:372@O idatm_type O2
atom id #4/A:372@CG idatm_type C3
atom id #4/A:372@CD idatm_type C3
atom id #4/A:372@NE idatm_type Ng+
atom id #4/A:372@NH1 idatm_type Ng+
atom id #4/A:372@NH2 idatm_type Ng+
atom id #4/A:372@CZ idatm_type C2
atom id #4/A:406@N idatm_type Npl
atom id #4/A:406@CA idatm_type C3
atom id #4/A:406@C idatm_type C2
atom id #4/A:406@CB idatm_type C3
atom id #4/A:406@O idatm_type O2
atom id #4/A:406@CG idatm_type Car
atom id #4/A:406@CD1 idatm_type Car
atom id #4/A:406@CD2 idatm_type Car
atom id #4/A:406@CE1 idatm_type Car
atom id #4/A:406@CE2 idatm_type Car
atom id #4/A:406@OH idatm_type O3
atom id #4/A:406@CZ idatm_type Car
> hide #!4 models
> select #2/A:120,153,229,294,372,406,
Expected an objects specifier or a keyword
> select #2/A:120,153,229,294,372,406
43 atoms, 40 bonds, 5 residues, 1 model selected
> show #!4 models
> ui tool show "Show Sequence Viewer"
> sequence chain #4/A
Alignment identifier is 4/A
> ui tool show "Show Sequence Viewer"
> sequence chain #2/A
Alignment identifier is 2/A
> ui tool show "Renumber Residues"
> renumber #2/A seqStart 83
388 residues renumbered
> select #2/A:120,153,229,294,372,406
63 atoms, 58 bonds, 6 residues, 1 model selected
> hide #!4 models
> show sel atoms
> color sel byhetero
> lighting full
> lighting simple
> lighting full
> save "/Users/iphan/Documents/ssgcid/Flu
> project/neuraminidase/test_rfdesign_protein100.png" width 831 height 739
> supersample 3
> save "/Users/iphan/Documents/ssgcid/Flu
> project/neuraminidase/neuraminidase.cxs"
——— End of log from Wed Dec 13 18:08:18 2023 ———
opened ChimeraX session
> hide #!2 models
> select add #2
3923 atoms, 4012 bonds, 488 residues, 1 model selected
> show #!1 models
> select subtract #2
1 model selected
> close #1-5
> open 6LXI format mmcif fromDatabase pdb
6lxi title:
Crystal structure of Z2B3 Fab in complex with influenza virus neuraminidase
from A/Brevig Mission/1/1918 (H1N1) [more info...]
Chain information for 6lxi #1
---
Chain | Description | UniProt
A B | Neuraminidase | NRAM_I18A0 1-469
C H | Heavy chain of Z2B3 Fab |
D L | Light chain of Z2B3 Fab |
Non-standard residues in 6lxi #1
---
BMA — beta-D-mannopyranose (beta-D-mannose; D-mannose; mannose)
CA — calcium ion
FUC — alpha-L-fucopyranose (alpha-L-fucose; 6-deoxy-alpha-L-galactopyranose;
L-fucose; fucose)
MAN — alpha-D-mannopyranose (alpha-D-mannose; D-mannose; mannose)
NAG — 2-acetamido-2-deoxy-beta-D-glucopyranose (N-acetyl-beta-D-glucosamine;
2-acetamido-2-deoxy-beta-D-glucose; 2-acetamido-2-deoxy-D-glucose;
2-acetamido-2-deoxy-glucose; N-ACETYL-D-GLUCOSAMINE)
6lxi mmCIF Assemblies
---
1| author_and_software_defined_assembly
2| author_and_software_defined_assembly
> select /C:69@CG2
1 atom, 1 residue, 1 model selected
> select up
7 atoms, 6 bonds, 1 residue, 1 model selected
> select up
42 atoms, 41 bonds, 6 residues, 1 model selected
> select up
1095 atoms, 1123 bonds, 145 residues, 1 model selected
> select up
1101 atoms, 1128 bonds, 146 residues, 1 model selected
> select up
1611 atoms, 1652 bonds, 214 residues, 1 model selected
> hide sel atoms
> show sel cartoons
> select /D:2@N
1 atom, 1 residue, 1 model selected
> select up
6 atoms, 5 bonds, 1 residue, 1 model selected
> select up
49 atoms, 49 bonds, 7 residues, 1 model selected
> select up
1108 atoms, 1135 bonds, 151 residues, 1 model selected
> select up
1112 atoms, 1138 bonds, 152 residues, 1 model selected
> select up
1325 atoms, 1357 bonds, 179 residues, 1 model selected
> hide sel atoms
> select /H:204@CG
1 atom, 1 residue, 1 model selected
> select up
7 atoms, 7 bonds, 1 residue, 1 model selected
> select up
73 atoms, 74 bonds, 10 residues, 1 model selected
> select up
1727 atoms, 1771 bonds, 232 residues, 1 model selected
> select up
1796 atoms, 1771 bonds, 301 residues, 1 model selected
> select up
12997 atoms, 12773 bonds, 2206 residues, 1 model selected
> select down
1796 atoms, 1771 bonds, 301 residues, 1 model selected
> hide sel atoms
> select /L:208@CE
1 atom, 1 residue, 1 model selected
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
52 atoms, 51 bonds, 7 residues, 1 model selected
> select up
1563 atoms, 1602 bonds, 211 residues, 1 model selected
> select up
1609 atoms, 1602 bonds, 257 residues, 1 model selected
> select up
12997 atoms, 12773 bonds, 2206 residues, 1 model selected
> select down
1609 atoms, 1602 bonds, 257 residues, 1 model selected
> hide sel atoms
> select /D:203@CA
1 atom, 1 residue, 1 model selected
> select up
4 atoms, 3 bonds, 1 residue, 1 model selected
> select up
43 atoms, 43 bonds, 6 residues, 1 model selected
> select up
122 atoms, 123 bonds, 17 residues, 1 model selected
> hide sel atoms
> select /A:389@CG2
1 atom, 1 residue, 1 model selected
> select up
7 atoms, 6 bonds, 1 residue, 1 model selected
> select up
92 atoms, 93 bonds, 11 residues, 1 model selected
> select up
2980 atoms, 3069 bonds, 387 residues, 1 model selected
> hide sel atoms
> select solvent
565 atoms, 565 residues, 1 model selected
> hide sel atoms
> select up
2 atoms, 1 bond, 1 residue, 1 model selected
> select up
10 atoms, 10 bonds, 1 residue, 1 model selected
> select up
60 atoms, 64 bonds, 5 residues, 1 model selected
> hide sel atoms
> select /A:501@CA
1 atom, 1 residue, 1 model selected
> select add /A:502@CA
2 atoms, 2 residues, 1 model selected
> hide sel atoms
> select /C:112
6 atoms, 5 bonds, 1 residue, 1 model selected
> select add /C:111
10 atoms, 8 bonds, 2 residues, 1 model selected
> select add /C:110
18 atoms, 15 bonds, 3 residues, 1 model selected
> select add /C:109
26 atoms, 22 bonds, 4 residues, 1 model selected
> select add /C:108
37 atoms, 32 bonds, 5 residues, 1 model selected
> select add /B:368@NH1
38 atoms, 32 bonds, 6 residues, 1 model selected
> select subtract /B:368@NH1
37 atoms, 32 bonds, 5 residues, 1 model selected
> select add /C:107
45 atoms, 39 bonds, 6 residues, 1 model selected
> select add /C:106
52 atoms, 45 bonds, 7 residues, 1 model selected
> select add /C:105
60 atoms, 52 bonds, 8 residues, 1 model selected
> select add /C:104
67 atoms, 59 bonds, 9 residues, 1 model selected
> select add /C:103
74 atoms, 65 bonds, 10 residues, 1 model selected
> select add /C:113
86 atoms, 77 bonds, 11 residues, 1 model selected
> select add /C:114
98 atoms, 89 bonds, 12 residues, 1 model selected
> color sel lime
> select /B:200@ND2
1 atom, 1 residue, 1 model selected
> select up
8 atoms, 7 bonds, 1 residue, 1 model selected
> select up
36 atoms, 36 bonds, 6 residues, 1 model selected
> select up
2980 atoms, 3069 bonds, 387 residues, 1 model selected
> select down
36 atoms, 36 bonds, 6 residues, 1 model selected
> select add /B:217@CD
37 atoms, 36 bonds, 7 residues, 1 model selected
> select up
45 atoms, 44 bonds, 7 residues, 1 model selected
> select up
91 atoms, 90 bonds, 13 residues, 1 model selected
> select up
2980 atoms, 3069 bonds, 387 residues, 1 model selected
> mlp sel
Map values for surface "6lxi_B SES surface": minimum -30.24, mean -5.764,
maximum 24.54
To also show corresponding color key, enter the above mlp command and add key
true
> transparency (#!1 & sel) 40
> hide #1.2 models
> hide #1.1 models
> hide sel atoms
> select /C:106
7 atoms, 6 bonds, 1 residue, 1 model selected
> select add /C:107
15 atoms, 13 bonds, 2 residues, 1 model selected
> select add /C:109
23 atoms, 20 bonds, 3 residues, 1 model selected
> select add /C:111
27 atoms, 23 bonds, 4 residues, 1 model selected
> select add /C:110
35 atoms, 30 bonds, 5 residues, 1 model selected
> select subtract /C:111
31 atoms, 27 bonds, 4 residues, 1 model selected
> select add /C:111
35 atoms, 30 bonds, 5 residues, 1 model selected
> select add /C:112
41 atoms, 35 bonds, 6 residues, 1 model selected
> select add /C:113
53 atoms, 47 bonds, 7 residues, 1 model selected
> select add /C:114
65 atoms, 59 bonds, 8 residues, 1 model selected
> select add /C:103
72 atoms, 65 bonds, 9 residues, 1 model selected
> select add /C:104
79 atoms, 72 bonds, 10 residues, 1 model selected
> select add /C:105
87 atoms, 79 bonds, 11 residues, 1 model selected
> select add /C:108
98 atoms, 89 bonds, 12 residues, 1 model selected
> color sel red
> select clear
> lighting full
> ui tool show "Side View"
> hide #!1 models
> show #!1 models
> select add #1.3
2980 atoms, 387 residues, 2 models selected
> coulombic sel
The following heavy (non-hydrogen) atoms are missing, which may result in
inaccurate electrostatics:
/B LYS 469 OXT
Using Amber 20 recommended default charges and atom types for standard
residues
Coulombic values for 6lxi_B SES surface #1.3: minimum, -16.06, mean -2.44,
maximum 9.17
To also show corresponding color key, enter the above coulombic command and
add key true
> select clear
> select add /C:114
12 atoms, 12 bonds, 1 residue, 1 model selected
> select add /C:113
24 atoms, 24 bonds, 2 residues, 1 model selected
> select add /B:399@CE3
25 atoms, 24 bonds, 3 residues, 1 model selected
> select subtract /B:399@CE3
24 atoms, 24 bonds, 2 residues, 2 models selected
> select add /C:112
30 atoms, 29 bonds, 3 residues, 1 model selected
> select add /C:111
34 atoms, 32 bonds, 4 residues, 1 model selected
> select add /C:110
42 atoms, 39 bonds, 5 residues, 1 model selected
> select add /C:109
50 atoms, 46 bonds, 6 residues, 1 model selected
> select add /C:108
61 atoms, 56 bonds, 7 residues, 1 model selected
> select add /C:107
69 atoms, 63 bonds, 8 residues, 1 model selected
> select add /C:106
76 atoms, 69 bonds, 9 residues, 1 model selected
> select add /C:105
84 atoms, 76 bonds, 10 residues, 1 model selected
> select add /C:104
91 atoms, 83 bonds, 11 residues, 1 model selected
> select add /C:103
98 atoms, 89 bonds, 12 residues, 1 model selected
> color sel lime
> select /B:467@O
1 atom, 1 residue, 1 model selected
> select clear
> select up
2 atoms, 1 bond, 1 residue, 1 model selected
> select up
11 atoms, 11 bonds, 1 residue, 1 model selected
> select up
60 atoms, 64 bonds, 5 residues, 1 model selected
> hide sel atoms
> select /C:114
12 atoms, 12 bonds, 1 residue, 1 model selected
> select add /C:113
24 atoms, 24 bonds, 2 residues, 1 model selected
> select add /C:112
30 atoms, 29 bonds, 3 residues, 1 model selected
> select add /C:111
34 atoms, 32 bonds, 4 residues, 1 model selected
> select add /C:110
42 atoms, 39 bonds, 5 residues, 1 model selected
> select add /B:118@NH1
43 atoms, 39 bonds, 6 residues, 1 model selected
> select subtract /B:118@NH1
42 atoms, 39 bonds, 5 residues, 2 models selected
> select add /C:108
53 atoms, 49 bonds, 6 residues, 1 model selected
> select add /C:109
61 atoms, 56 bonds, 7 residues, 1 model selected
> select add /C:107
69 atoms, 63 bonds, 8 residues, 1 model selected
> select add /C:106
76 atoms, 69 bonds, 9 residues, 1 model selected
> select add /B:399@CZ3
77 atoms, 69 bonds, 10 residues, 1 model selected
> select subtract /B:399@CZ3
76 atoms, 69 bonds, 9 residues, 2 models selected
> select add /C:105
84 atoms, 76 bonds, 10 residues, 1 model selected
> select add /C:104
91 atoms, 83 bonds, 11 residues, 1 model selected
> select add /C:103
98 atoms, 89 bonds, 12 residues, 1 model selected
> show sel atoms
> style sel ball
Changed 98 atom styles
> color sel byhetero
> select clear
> select /C:114@CA
1 atom, 1 residue, 1 model selected
> select up
12 atoms, 12 bonds, 1 residue, 1 model selected
> hide sel atoms
> ui tool show "Color Actions"
[Repeated 1 time(s)]
> color sel plum
> select clear
> save "/Users/iphan/Documents/ssgcid/Flu
> project/documents/neuraminidase_svtd.cxs"
——— End of log from Wed May 1 10:43:48 2024 ———
opened ChimeraX session
> select /C:58
7 atoms, 6 bonds, 1 residue, 1 model selected
> select up
34 atoms, 34 bonds, 4 residues, 1 model selected
> select up
1095 atoms, 1123 bonds, 145 residues, 1 model selected
> ui tool show "Show Sequence Viewer"
> sequence chain /A /B
Alignment identifier is 1
> sequence chain /H /C
Alignment identifier is 2
> interfaces select /C & ::polymer_type>0 contacting /B & ::polymer_type>0
> areaCutoff 0 bothSides true
30 contacting residues
> hide #1.3.1 models
> hide #!1.3 models
> show #1.3 models
> select add #1.3
3099 atoms, 402 residues, 3 models selected
> select subtract #1.3
119 atoms, 15 residues, 2 models selected
> undo
[Repeated 1 time(s)]
> toolshed show
> ui tool show Log
> select up
944 atoms, 960 bonds, 124 residues, 2 models selected
> select down
250 atoms, 30 residues, 2 models selected
> select intersect sideonly
59 atoms, 15 residues, 1 model selected
> hide #!1.3 models
> show #1.2 models
> hide #1.2 models
> show #1.3 models
> hide #1.3.1 models
> hide #!1.3 models
> hide #!1 models
> show #!1 models
> show #1.3 models
> select intersect /C
59 atoms, 15 residues, 1 model selected
> show sel cartoons
> hide #!1.3 models
> show #1.3 models
> hide #!1.3 models
> select /C
1657 atoms, 1652 bonds, 2 pseudobonds, 260 residues, 2 models selected
> show #1.3 models
> select /B
3147 atoms, 3069 bonds, 13 pseudobonds, 554 residues, 2 models selected
> show sel cartoons
> hide #!1.3 models
> select subtract #1.3
167 atoms, 3 pseudobonds, 167 residues, 3 models selected
> select add #1.1
167 atoms, 25 pseudobonds, 167 residues, 2 models selected
> select add #1
12997 atoms, 12773 bonds, 29 pseudobonds, 2206 residues, 3 models selected
> select subtract #1
1 model selected
> interfaces select /C & ::polymer_type>0 contacting /B & ::polymer_type>0
> areaCutoff 0 bothSides true
30 contacting residues
> select intersect sideonly
59 atoms, 15 residues, 1 model selected
> undo
[Repeated 1 time(s)]
> interfaces select /B & ::polymer_type>0 contacting /C & ::polymer_type>0
> areaCutoff 0 bothSides true
30 contacting residues
> select intersect sideonly
59 atoms, 15 residues, 1 model selected
> undo
> select clear
> interfaces select /B & ::polymer_type>0 contacting /C & ::polymer_type>0
> areaCutoff 0 bothSides true
30 contacting residues
> show sel atoms
> select up
944 atoms, 960 bonds, 124 residues, 2 models selected
> select down
250 atoms, 30 residues, 2 models selected
> style sel stick
Changed 250 atom styles
> select up
944 atoms, 960 bonds, 124 residues, 2 models selected
> select down
250 atoms, 30 residues, 2 models selected
> color (#!1 & sel) red
> undo
> select intersect /B
Nothing selected
> interfaces select /B & ::polymer_type>0 contacting /C & ::polymer_type>0
> areaCutoff 0 bothSides true
30 contacting residues
> select subtract /C/H
131 atoms, 15 residues, 2 models selected
> select up
673 atoms, 683 bonds, 89 residues, 2 models selected
> select down
131 atoms, 15 residues, 2 models selected
> color (#!1 & sel) red
> color (#!1 & sel) yellow
> set bgColor black
> select intersect sidechain
Nothing selected
> undo
> select clear
> interfaces select /B & ::polymer_type>0 contacting /C & ::polymer_type>0
> areaCutoff 0 bothSides true
30 contacting residues
> select subtract backbone
130 atoms, 30 residues, 2 models selected
> undo
[Repeated 5 time(s)]
> select clear
> interfaces select /B & ::polymer_type>0 contacting /C & ::polymer_type>0
> areaCutoff 0 bothSides true
30 contacting residues
> select subtract /C
131 atoms, 15 residues, 2 models selected
> select subtract backbone
71 atoms, 15 residues, 2 models selected
> color (#!1 & sel) byhetero
> color (#!1 & sel) light gray
> undo
> select subtract backbone
71 atoms, 15 residues, 2 models selected
> select subtract backbone
71 atoms, 15 residues, 2 models selected
> color (#!1 & sel) light gray
> color (#!1 & sel) byhetero
> style sel sphere
Changed 71 atom styles
> style sel sphere
Changed 71 atom styles
> style sel sphere
Changed 71 atom styles
> style sel ball
Changed 71 atom styles
> style sel stick
Changed 71 atom styles
> label (#!1 & sel) text "{0.label_one_letter_code}
> {0.number}{0.insertion_code}"
> select clear
> undo
> list residues sel
Unknown command: list residues sel
> list residues
Unknown command: list residues
> list re
Unknown command: list re
> list res
Unknown command: list res
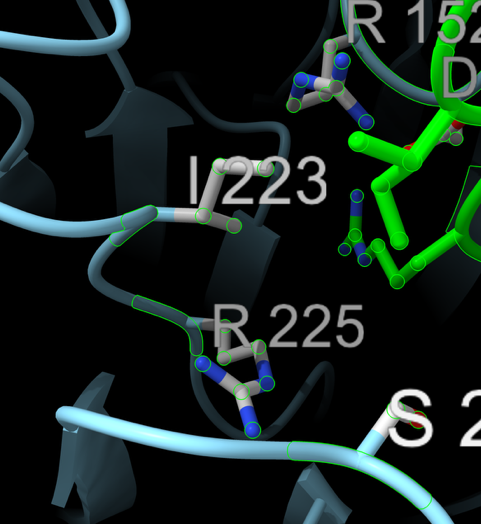
> info residues sel
residue id /B:118 name ARG index 117
residue id /B:149 name VAL index 148
residue id /B:150 name LYS index 149
residue id /B:151 name ASP index 150
residue id /B:152 name ARG index 151
residue id /B:223 name ILE index 222
residue id /B:225 name ARG index 224
residue id /B:247 name SER index 246
residue id /B:344 name ASN index 343
residue id /B:368 name ARG index 367
residue id /B:430 name GLN index 429
residue id /B:431 name PRO index 430
residue id /B:432 name LYS index 431
residue id /B:434 name ASN index 433
residue id /B:436 name ILE index 435
> select /C
1657 atoms, 1652 bonds, 2 pseudobonds, 260 residues, 2 models selected
> select /B
3147 atoms, 3069 bonds, 13 pseudobonds, 554 residues, 2 models selected
> select intersect backbone
1547 bonds, 1 model selected
> ui tool show "Color Actions"
> select clear
> select /B
3147 atoms, 3069 bonds, 13 pseudobonds, 554 residues, 2 models selected
> select subtract sideonly
1715 atoms, 1547 bonds, 8 pseudobonds, 554 residues, 3 models selected
> color (#!1 & sel) cornflower blue
> ui tool show "Color Actions"
> color sel sky blue
> select clear
OpenGL version: 4.1 Metal - 88.1
OpenGL renderer: Apple M1 Pro
OpenGL vendor: Apple
Python: 3.11.4
Locale: UTF-8
Qt version: PyQt6 6.7.1, Qt 6.7.1
Qt runtime version: 6.7.3
Qt platform: cocoa
Hardware:
Hardware Overview:
Model Name: MacBook Pro
Model Identifier: MacBookPro18,3
Model Number: Z15G001WALL/A
Chip: Apple M1 Pro
Total Number of Cores: 8 (6 performance and 2 efficiency)
Memory: 32 GB
System Firmware Version: 11881.81.2
OS Loader Version: 10151.140.19.700.2
Software:
System Software Overview:
System Version: macOS 14.7.3 (23H417)
Kernel Version: Darwin 23.6.0
Time since boot: 2 days, 8 hours
Graphics/Displays:
Apple M1 Pro:
Chipset Model: Apple M1 Pro
Type: GPU
Bus: Built-In
Total Number of Cores: 14
Vendor: Apple (0x106b)
Metal Support: Metal 3
Displays:
Color LCD:
Display Type: Built-in Liquid Retina XDR Display
Resolution: 3024 x 1964 Retina
Main Display: Yes
Mirror: Off
Online: Yes
Automatically Adjust Brightness: No
Connection Type: Internal
Installed Packages:
alabaster: 1.0.0
anyio: 4.7.0
appdirs: 1.4.4
appnope: 0.1.4
asttokens: 3.0.0
auditwheel: 6.1.0
babel: 2.16.0
beautifulsoup4: 4.12.3
blockdiag: 3.0.0
blosc2: 3.0.0
build: 1.2.1
certifi: 2023.11.17
cftime: 1.6.4.post1
charset-normalizer: 3.4.0
ChimeraX-AddCharge: 1.5.18
ChimeraX-AddH: 2.2.6
ChimeraX-AlignmentAlgorithms: 2.0.2
ChimeraX-AlignmentHdrs: 3.5
ChimeraX-AlignmentMatrices: 2.1
ChimeraX-Alignments: 2.16.1
ChimeraX-AlphaFold: 1.0.1
ChimeraX-AltlocExplorer: 1.1.2
ChimeraX-AmberInfo: 1.0
ChimeraX-Arrays: 1.1
ChimeraX-Atomic: 1.58.8
ChimeraX-AtomicLibrary: 14.1.11
ChimeraX-AtomSearch: 2.0.1
ChimeraX-AxesPlanes: 2.4
ChimeraX-BasicActions: 1.1.2
ChimeraX-BILD: 1.0
ChimeraX-BlastProtein: 3.0.0
ChimeraX-BondRot: 2.0.4
ChimeraX-BugReporter: 1.0.1
ChimeraX-BuildStructure: 2.13.1
ChimeraX-Bumps: 1.0
ChimeraX-BundleBuilder: 1.4.0
ChimeraX-ButtonPanel: 1.0.1
ChimeraX-CageBuilder: 1.0.1
ChimeraX-CellPack: 1.0
ChimeraX-Centroids: 1.4
ChimeraX-ChangeChains: 1.1
ChimeraX-CheckWaters: 1.4
ChimeraX-ChemGroup: 2.0.1
ChimeraX-Clashes: 2.3
ChimeraX-ColorActions: 1.0.5
ChimeraX-ColorGlobe: 1.0
ChimeraX-ColorKey: 1.5.6
ChimeraX-CommandLine: 1.2.5
ChimeraX-ConnectStructure: 2.0.1
ChimeraX-Contacts: 1.0.1
ChimeraX-Core: 1.9
ChimeraX-CoreFormats: 1.2
ChimeraX-coulombic: 1.4.4
ChimeraX-Crosslinks: 1.0
ChimeraX-Crystal: 1.0
ChimeraX-CrystalContacts: 1.0.1
ChimeraX-DataFormats: 1.2.3
ChimeraX-Dicom: 1.2.6
ChimeraX-DistMonitor: 1.4.2
ChimeraX-DockPrep: 1.1.3
ChimeraX-Dssp: 2.0
ChimeraX-EMDB-SFF: 1.0
ChimeraX-ESMFold: 1.0
ChimeraX-FileHistory: 1.0.1
ChimeraX-FunctionKey: 1.0.1
ChimeraX-Geometry: 1.3
ChimeraX-gltf: 1.0
ChimeraX-Graphics: 1.4.1
ChimeraX-Hbonds: 2.5
ChimeraX-Help: 1.3
ChimeraX-HKCage: 1.3
ChimeraX-IHM: 1.1
ChimeraX-ImageFormats: 1.2
ChimeraX-IMOD: 1.0
ChimeraX-IO: 1.0.3
ChimeraX-ItemsInspection: 1.0.1
ChimeraX-IUPAC: 1.0
ChimeraX-KVFinder: 1.2.1
ChimeraX-Label: 1.1.14
ChimeraX-ListInfo: 1.2.2
ChimeraX-Log: 1.2
ChimeraX-LookingGlass: 1.1
ChimeraX-Maestro: 1.9.1
ChimeraX-Map: 1.3
ChimeraX-MapData: 2.0
ChimeraX-MapEraser: 1.0.1
ChimeraX-MapFilter: 2.0.1
ChimeraX-MapFit: 2.0
ChimeraX-MapSeries: 2.1.1
ChimeraX-Markers: 1.0.1
ChimeraX-Mask: 1.0.2
ChimeraX-MatchMaker: 2.1.6
ChimeraX-MCopy: 1.0
ChimeraX-MDcrds: 2.7.2
ChimeraX-MedicalToolbar: 1.1
ChimeraX-Meeting: 1.0.1
ChimeraX-MLP: 1.1.1
ChimeraX-mmCIF: 2.14.2
ChimeraX-MMTF: 2.2
ChimeraX-ModelArchive: 1.0
ChimeraX-Modeller: 1.5.18
ChimeraX-ModelPanel: 1.5
ChimeraX-ModelSeries: 1.0.1
ChimeraX-Mol2: 2.0.3
ChimeraX-Mole: 1.0
ChimeraX-Morph: 1.0.2
ChimeraX-MouseModes: 1.2
ChimeraX-Movie: 1.0
ChimeraX-MutationScores: 1.0
ChimeraX-Neuron: 1.0
ChimeraX-Nifti: 1.2
ChimeraX-NMRSTAR: 1.0.2
ChimeraX-NRRD: 1.2
ChimeraX-Nucleotides: 2.0.3
ChimeraX-OpenCommand: 1.14
ChimeraX-OrthoPick: 1.0.1
ChimeraX-PDB: 2.7.6
ChimeraX-PDBBio: 1.0.1
ChimeraX-PDBLibrary: 1.0.4
ChimeraX-PDBMatrices: 1.0
ChimeraX-PickBlobs: 1.0.1
ChimeraX-Positions: 1.0
ChimeraX-PresetMgr: 1.1.2
ChimeraX-PubChem: 2.2
ChimeraX-ReadPbonds: 1.0.1
ChimeraX-Registration: 1.1.2
ChimeraX-RemoteControl: 1.0
ChimeraX-RenderByAttr: 1.6.2
ChimeraX-RenumberResidues: 1.1
ChimeraX-ResidueFit: 1.0.1
ChimeraX-RestServer: 1.3.1
ChimeraX-RNALayout: 1.0
ChimeraX-RotamerLibMgr: 4.0
ChimeraX-RotamerLibsDunbrack: 2.0
ChimeraX-RotamerLibsDynameomics: 2.0
ChimeraX-RotamerLibsRichardson: 2.0
ChimeraX-SaveCommand: 1.5.1
ChimeraX-SchemeMgr: 1.0
ChimeraX-SDF: 2.0.2
ChimeraX-Segger: 1.0
ChimeraX-Segment: 1.0.1
ChimeraX-Segmentations: 3.5.6
ChimeraX-SelInspector: 1.0
ChimeraX-SeqView: 2.14
ChimeraX-Shape: 1.0.1
ChimeraX-Shell: 1.0.1
ChimeraX-Shortcuts: 1.2.0
ChimeraX-ShowSequences: 1.0.3
ChimeraX-SideView: 1.0.1
ChimeraX-SimilarStructures: 1.0.1
ChimeraX-Smiles: 2.1.2
ChimeraX-SmoothLines: 1.0
ChimeraX-SpaceNavigator: 1.0
ChimeraX-StdCommands: 1.18.1
ChimeraX-STL: 1.0.1
ChimeraX-Storm: 1.0
ChimeraX-StructMeasure: 1.2.1
ChimeraX-Struts: 1.0.1
ChimeraX-Surface: 1.0.1
ChimeraX-SwapAA: 2.0.1
ChimeraX-SwapRes: 2.5
ChimeraX-TapeMeasure: 1.0
ChimeraX-TaskManager: 1.0
ChimeraX-Test: 1.0
ChimeraX-Toolbar: 1.2.3
ChimeraX-ToolshedUtils: 1.2.4
ChimeraX-Topography: 1.0
ChimeraX-ToQuest: 1.0
ChimeraX-Tug: 1.0.1
ChimeraX-UI: 1.41
ChimeraX-Umap: 1.0
ChimeraX-uniprot: 2.3.1
ChimeraX-UnitCell: 1.0.1
ChimeraX-ViewDockX: 1.4.4
ChimeraX-VIPERdb: 1.0
ChimeraX-Vive: 1.1
ChimeraX-VolumeMenu: 1.0.1
ChimeraX-vrml: 1.0
ChimeraX-VTK: 1.0
ChimeraX-WavefrontOBJ: 1.0
ChimeraX-WebCam: 1.0.2
ChimeraX-WebServices: 1.1.4
ChimeraX-Zone: 1.0.1
colorama: 0.4.6
comm: 0.2.2
contourpy: 1.3.1
cxservices: 1.2.3
cycler: 0.12.1
Cython: 3.0.10
debugpy: 1.8.9
decorator: 5.1.1
docutils: 0.21.2
executing: 2.1.0
filelock: 3.15.4
fonttools: 4.55.3
funcparserlib: 2.0.0a0
glfw: 2.8.0
grako: 3.16.5
h11: 0.14.0
h5py: 3.12.1
html2text: 2024.2.26
httpcore: 1.0.7
httpx: 0.28.1
idna: 3.10
ihm: 1.3
imagecodecs: 2024.6.1
imagesize: 1.4.1
ipykernel: 6.29.5
ipython: 8.26.0
ipywidgets: 8.1.5
jedi: 0.19.1
Jinja2: 3.1.4
jupyter_client: 8.6.2
jupyter_core: 5.7.2
jupyterlab_widgets: 3.0.13
kiwisolver: 1.4.7
line_profiler: 4.1.3
lxml: 5.2.2
lz4: 4.3.3
MarkupSafe: 3.0.2
matplotlib: 3.9.2
matplotlib-inline: 0.1.7
msgpack: 1.0.8
ndindex: 1.9.2
nest-asyncio: 1.6.0
netCDF4: 1.6.5
networkx: 3.3
nibabel: 5.2.0
nptyping: 2.5.0
numexpr: 2.10.2
numpy: 1.26.4
openvr: 1.26.701
packaging: 23.2
ParmEd: 4.2.2
parso: 0.8.4
pep517: 0.13.1
pexpect: 4.9.0
pillow: 10.4.0
pip: 24.2
pkginfo: 1.11.1
platformdirs: 4.3.6
prompt_toolkit: 3.0.48
psutil: 6.0.0
ptyprocess: 0.7.0
pure_eval: 0.2.3
py-cpuinfo: 9.0.0
pycollada: 0.8
pydicom: 2.4.4
pyelftools: 0.31
Pygments: 2.18.0
pynmrstar: 3.3.4
pynrrd: 1.0.0
PyOpenGL: 3.1.7
PyOpenGL-accelerate: 3.1.7
pyopenxr: 1.0.3401
pyparsing: 3.2.0
pyproject_hooks: 1.2.0
PyQt6-commercial: 6.7.1
PyQt6-Qt6: 6.7.3
PyQt6-WebEngine-commercial: 6.7.0
PyQt6-WebEngine-Qt6: 6.7.3
PyQt6-WebEngineSubwheel-Qt6: 6.7.3
PyQt6_sip: 13.8.0
python-dateutil: 2.9.0.post0
pytz: 2024.2
pyzmq: 26.2.0
qtconsole: 5.5.2
QtPy: 2.4.2
qtshim: 1.0
RandomWords: 0.4.0
requests: 2.32.3
scipy: 1.14.0
setuptools: 72.1.0
sfftk-rw: 0.8.1
six: 1.16.0
sniffio: 1.3.1
snowballstemmer: 2.2.0
sortedcontainers: 2.4.0
soupsieve: 2.6
Sphinx: 8.0.2
sphinx-autodoc-typehints: 2.2.3
sphinxcontrib-applehelp: 2.0.0
sphinxcontrib-blockdiag: 3.0.0
sphinxcontrib-devhelp: 2.0.0
sphinxcontrib-htmlhelp: 2.1.0
sphinxcontrib-jsmath: 1.0.1
sphinxcontrib-qthelp: 2.0.0
sphinxcontrib-serializinghtml: 2.0.0
stack-data: 0.6.3
superqt: 0.6.3
tables: 3.10.1
tcia_utils: 1.5.1
tifffile: 2024.7.24
tinyarray: 1.2.4
tornado: 6.4.2
traitlets: 5.14.3
typing_extensions: 4.12.2
tzdata: 2024.2
urllib3: 2.2.3
wcwidth: 0.2.13
webcolors: 24.6.0
wheel: 0.43.0
wheel-filename: 1.4.1
widgetsnbextension: 4.0.13
Attachments (3)
{kind=link}
Change History (22)
comment:1 by , 18 months ago
| Component: | Unassigned → General Controls |
|---|---|
| Owner: | set to |
| Platform: | → all |
| Project: | → ChimeraX |
| Status: | new → accepted |
| Summary: | ChimeraX bug report submission → Selection intersection problem |
comment:2 by , 18 months ago
| Status: | accepted → feedback |
|---|
comment:3 by , 18 months ago
Hello Eric, I've been having this problem with multiple structures. The current one is 6lxi. I see the sidechains, and I see that they are selected. I think the problem is that I am starting with 'Contacts'. That selects ATOMS and not bonds: [cid:ac62d4c8-e715-4749-8f45-936b511f5ea7@namprd08.prod.outlook.com] I thought perhaps the Intersect does not work because the selection is missing the bonds. So I select 'expand' to include the bonds. Result = it selects everything in the same secondary structure element, including residues that are not making any contacts with the other chain. action=select Contacts; results=select atoms making contacts = OK [cid:8b3a9825-aa4d-4919-885d-b400c05de540@namprd08.prod.outlook.com] action=Expand; expected results=select residues including bonds between selected atoms; actual result=selects secondary structure elements [cid:b71428f2-1655-4d6e-a4b6-3597e752058b@namprd08.prod.outlook.com] So I cannot use Expand to select the bonds. Unfortunately, when I start with the ATOM selection, the Intersect action does not work. Isabelle > > > > > >

by , 18 months ago
| Attachment: | =?utf-8?B?U2NyZWVuc2hvdCAyMDI1LTAyLTA3IGF0IDEwLjI4LjUy4oCvQU0ucG5n?= added |
|---|
Added by email2trac
by , 18 months ago
| Attachment: | =?utf-8?B?U2NyZWVuc2hvdCAyMDI1LTAyLTA3IGF0IDEwLjI5LjA34oCvQU0ucG5n?= added |
|---|
Added by email2trac
comment:4 by , 18 months ago
| Status: | feedback → accepted |
|---|
comment:5 by , 18 months ago
| Cc: | added |
|---|---|
| Component: | General Controls → Structure Analysis |
| Owner: | changed from to |
| Status: | accepted → assigned |
Tom, is there a reason that "interfaces select" selects only atoms and not the connecting bonds?
comment:6 by , 18 months ago
Isabelle, as a temporary workaround, after selecting the chain contacts the command "sel sel-residues" will get the bonds selected also.
comment:7 by , 18 months ago
>the command "sel sel-residues" will get the bonds selected also. Ooh brilliant, that worked. Thanks a bunch! Isabelle
comment:8 by , 18 months ago
Eric, I'm not sure why "interfaces select" only selects the atoms and not the bonds. I guess it is because the interfaces are defined by contacts which are between the atoms. I'm baffled how that would cause "select intersect" not to work. Also the log of this bug report shows behavior that I do not get in my ChimeraX 1.9. For instance the log in this report shows
open 6hg0
select #1:119,120,135,152,153,181,227,228,229,279,294,351,372,406
130 atoms, 121 bonds, 14 residues, 1 model selected
select intersect sideonly
61 bonds, 1 model selected
Why didn't the atoms get selected in the intersection. When I do these commands in ChimeraX 1.9 they do get selected.
open 6hg0
select #1:119,120,135,152,153,181,227,228,229,279,294,351,372,406
130 atoms, 121 bonds, 14 residues, 1 model selected
select intersect sideonly
74 atoms, 61 bonds, 14 residues, 1 model selected
My only idea about how that could happen is that the "select #1:119..." was actually logged but not executed, and some tool did a selection that wasn't the same as what that command produces.
It would help if the reporter of this bug gave a simple step by step example with an actual PDB file since the description written in the bug report doesn't reproduce the problem for me.
comment:9 by , 18 months ago
As per comment #3, I think the complaint/problem here is that the bonds aren't selected, not that there is no selection at all.
So it seems to me like the correct fix is for "interfaces select" to also select bonds. It is documented to "select interface residues", which would certainly seem like bonds too. But if there is some real reason that the command doesn't select bonds, I can work around that by having the selection menu issue an additional "sel sel-residues" afterward. But that seems kind of kludgey compared to just having "interfaces select" select the bonds itself.
comment:10 by , 18 months ago
I agree, it is pretty bizarre to select the atoms and not the bonds.
But I am mystified by the Log in this report that shows "select intersect sideonly" selecting just the bonds, and when I try the same commands selects the atoms. I'm not inclined to fix anything until I can reproduce the problem. There is clearly something I don't understand.
comment:11 by , 18 months ago
I can "sort of" reproduce by going to the Python shell and deselecting the atoms only in between the commands. But according to the log the Python shell had not been started, and I can't think of any way to deselect atoms only without also putting something in the log.
comment:12 by , 18 months ago
Isabelle, could you try a simple test in your ChimeraX 1.9 running these 3 commands and copy and paste the log output?
open 6hg0
select #1:119,120,135,152,153,181,227,228,229,279,294,351,372,406
select intersect sideonly
These are 3 commands I see from your bug report and in the bug report it says no atoms are end up selected. But for me the atoms do get selected. I'm guessing you did not type that second command in your original ChimeraX session with the dozen residue numbers and instead you pushed some button that generated that command. It looks like the residues near the ligand. Could you tell me if you recall what button you pressed to select those residues?
I know this problem is pretty minor compared to a lot of our grant money being yanked back. But this one we can easily fix if we know how to reproduce it. Thanks!
comment:13 by , 18 months ago
This is the workflow: - open 6lxi - Menu: Select > Contacts... select contacts of: chain B with Chain C => result: 30 residues selected, 15 on each chain - Menu Mode: Intersect (-) - Menu Select (-) > Structure > Sidechain Only => result: all the residues in Chain B are de-selected. Log: interfaces select #1/B & ::polymer_type>0 contacting #1/C & ::polymer_type>0 areaCutoff 0 bothSides true 30 contacting residues select intersect sideonly 59 atoms, 15 residues, 1 model selected What I expected: side-chains of the 30 contacting residues on the 2 models selected. Isabelle
comment:14 by , 18 months ago
Thanks Isabelle! When I do exactly your example in ChimeraX 1.9 on Mac I get both chain B and C residues selected. Here is the Log output I get with 30 residues selected after the intersection.
interfaces select /B & ::polymer_type>0 contacting /C & ::polymer_type>0 areaCutoff 0 bothSides true
30 contacting residues
select intersect sideonly
130 atoms, 30 residues, 1 model selected
But I notice another difference beside the end result. Your "interfaces select" command has #1/B and #1/C while mine has /B and /C. I believe that means you have some other model open. That shouldn't make a difference but maybe that extra model is needed to reproduce this bug. Can you tell me if there is another model open and what it is? And can you try in a fresh ChimeraX session with no other model open?
comment:15 by , 18 months ago
Crickey, you are right! So I had another model open that only contained chains B and C that I saved in ChimeraX from PDB 6lxi After I restarted a fresh session and only loaded the PDB file, the "intersect sideonly" worked as expected. Weird! Isabelle
comment:16 by , 18 months ago
It looks like my issue was a corrupted session. I just added the model with only the B/C chains, and "intersect sideonly" still works fine. One of those mysteries \U0001f937\U0001f3fb\u200d\u2640\ufe0f I.
comment:17 by , 18 months ago
Good. If you want to send the session with this weird problem we could look at it. Or we can call it done and if you see the problem again report it again.
comment:18 by , 18 months ago
I wouldn't normally say this, but in the current circumstances I very much hope I will be able to report the problem again if it arises.
I.
Hi Isabelle,
--Eric