Opened 3 years ago
Closed 10 months ago
#9387 closed task (fixed)
ESPALOMA-based parameter estimation
| Reported by: | Tristan Croll | Owned by: | Eric Pettersen |
|---|---|---|---|
| Priority: | normal | Milestone: | |
| Component: | Structure Editing | Version: | |
| Keywords: | Cc: | Tom Goddard, Elaine Meng | |
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
The following bug report has been submitted:
Platform: Linux-5.19.0-43-generic-x86_64-with-glibc2.35
ChimeraX Version: 1.6.1 (2023-05-09 17:57:07 UTC)
Description
I've been talking quite a bit lately with John Chodera about ESPALOMA (https://arxiv.org/abs/2307.07085), which is apparently now outperforming AMBER in terms of RMS energy and force error for protein, RNA and small molecules, while doing away entirely with the use of residue templates. It seems like a natural path forward for interactive MD methods like ISOLDE, given that it naturally handles new organic ligands and post-translational modifications without any extra effort from the user.
Apart from the (possibly substantial) challenge of porting it from conda, probably the biggest issue is that in its current incarnation the input (a graph representation of the molecule(s) with elements as nodes and bonds as edges) requires explicit specification of bond orders, and ChimeraX doesn't always get those right. For example, if I take something with alternating single/double bonds like the carotenoid V7N and save it to .mol2 from ChimeraX, then the intervening single bonds are erroneously written as double. A secondary issue is that it also requires overall net charge to be specified, something that ChimeraX also doesn't always get correct (see #9174).
The aim of this ticket is to discuss ways to successfully use the ESPALOMA framework to efficiently and accurately parameterise a model that's open in ChimeraX. One attractive idea I've discussed with John is to do a ChimeraX-specific retraining where the node labels are replaced with ChimeraX `idatm_type`s with the edges being simple generic bonds - in theory I think that should be enough for it to converge to a similar-quality solution, and the actual training apparently isn't hugely resource-intensive.
Log:
> isolde shorthand
Initialising ISOLDE-specific command aliases:
Alias Equivalent full command
-------------------------------------------------
st isolde step {arguments}
aw isolde add water {arguments}
awsf isolde add water {arguments} sim false
al isolde add ligand {arguments}
aa isolde add aa $1 sel {arguments}
ht isolde mod his sel {arguments}
so setattr sel atoms occupancy {arguments}
ab isolde adjust bfactors {arguments}
ss isolde sim start sel
rt isolde release torsions sel {arguments}
rd isolde release distances sel {arguments}
ra rd; rt
pf isolde pepflip sel
cf isolde cisflip sel
cbb color bfactor {arguments}
cbo color byattr occupancy {arguments}
cbc color {arguments} bychain; color {arguments} byhet
cs clipper set contourSensitivity {arguments}
UCSF ChimeraX version: 1.6.1 (2023-05-09)
© 2016-2023 Regents of the University of California. All rights reserved.
How to cite UCSF ChimeraX
> open /home/tcroll/Downloads/V7N_model.sdf
> save v7n.mol2 #1
OpenGL version: 3.3.0 NVIDIA 515.105.01
OpenGL renderer: NVIDIA GeForce RTX 3070/PCIe/SSE2
OpenGL vendor: NVIDIA Corporation
Python: 3.9.11
Locale: en_GB.UTF-8
Qt version: PyQt6 6.4.2, Qt 6.4.2
Qt runtime version: 6.4.3
Qt platform: xcb
XDG_SESSION_TYPE=x11
DESKTOP_SESSION=ubuntu
XDG_SESSION_DESKTOP=ubuntu
XDG_CURRENT_DESKTOP=ubuntu:GNOME
DISPLAY=:1
Manufacturer: Dell Inc.
Model: XPS 8950
OS: Ubuntu 22.04 Jammy Jellyfish
Architecture: 64bit ELF
Virtual Machine: none
CPU: 20 12th Gen Intel(R) Core(TM) i7-12700
Cache Size: 25600 KB
Memory:
total used free shared buff/cache available
Mem: 31Gi 16Gi 4.5Gi 820Mi 10Gi 13Gi
Swap: 2.0Gi 2.0Gi 2.0Mi
Graphics:
0000:01:00.0 VGA compatible controller [0300]: NVIDIA Corporation GA104 [GeForce RTX 3070 Lite Hash Rate] [10de:2488] (rev a1)
Subsystem: Dell GA104 [GeForce RTX 3070 Lite Hash Rate] [1028:c903]
Kernel driver in use: nvidia
Installed Packages:
alabaster: 0.7.13
appdirs: 1.4.4
asttokens: 2.2.1
Babel: 2.12.1
backcall: 0.2.0
beautifulsoup4: 4.11.2
blockdiag: 3.0.0
build: 0.10.0
certifi: 2023.5.7
cftime: 1.6.2
charset-normalizer: 3.1.0
ChimeraX-AddCharge: 1.5.9.1
ChimeraX-AddH: 2.2.5
ChimeraX-AlignmentAlgorithms: 2.0.1
ChimeraX-AlignmentHdrs: 3.3.1
ChimeraX-AlignmentMatrices: 2.1
ChimeraX-Alignments: 2.9.3
ChimeraX-AlphaFold: 1.0
ChimeraX-AltlocExplorer: 1.0.3
ChimeraX-AmberInfo: 1.0
ChimeraX-Arrays: 1.1
ChimeraX-Atomic: 1.43.10
ChimeraX-AtomicLibrary: 10.0.6
ChimeraX-AtomSearch: 2.0.1
ChimeraX-AxesPlanes: 2.3.2
ChimeraX-BasicActions: 1.1.2
ChimeraX-BILD: 1.0
ChimeraX-BlastProtein: 2.1.2
ChimeraX-BondRot: 2.0.1
ChimeraX-BugReporter: 1.0.1
ChimeraX-BuildStructure: 2.8
ChimeraX-Bumps: 1.0
ChimeraX-BundleBuilder: 1.2.2
ChimeraX-ButtonPanel: 1.0.1
ChimeraX-CageBuilder: 1.0.1
ChimeraX-CellPack: 1.0
ChimeraX-Centroids: 1.3.2
ChimeraX-ChangeChains: 1.0.2
ChimeraX-CheckWaters: 1.3.1
ChimeraX-ChemGroup: 2.0.1
ChimeraX-Clashes: 2.2.4
ChimeraX-Clipper: 0.21.0
ChimeraX-ColorActions: 1.0.3
ChimeraX-ColorGlobe: 1.0
ChimeraX-ColorKey: 1.5.3
ChimeraX-CommandLine: 1.2.5
ChimeraX-ConnectStructure: 2.0.1
ChimeraX-Contacts: 1.0.1
ChimeraX-Core: 1.6.1
ChimeraX-CoreFormats: 1.1
ChimeraX-coulombic: 1.4.2
ChimeraX-Crosslinks: 1.0
ChimeraX-Crystal: 1.0
ChimeraX-CrystalContacts: 1.0.1
ChimeraX-DataFormats: 1.2.3
ChimeraX-Dicom: 1.2
ChimeraX-DistMonitor: 1.4
ChimeraX-DockPrep: 1.1.1
ChimeraX-Dssp: 2.0
ChimeraX-EMDB-SFF: 1.0
ChimeraX-ESMFold: 1.0
ChimeraX-FileHistory: 1.0.1
ChimeraX-FunctionKey: 1.0.1
ChimeraX-Geometry: 1.3
ChimeraX-gltf: 1.0
ChimeraX-Graphics: 1.1.1
ChimeraX-Hbonds: 2.4
ChimeraX-Help: 1.2.1
ChimeraX-HKCage: 1.3
ChimeraX-IHM: 1.1
ChimeraX-ImageFormats: 1.2
ChimeraX-IMOD: 1.0
ChimeraX-IO: 1.0.1
ChimeraX-ISOLDE: 1.6.0
ChimeraX-ItemsInspection: 1.0.1
ChimeraX-Label: 1.1.7
ChimeraX-LinuxSupport: 1.0.1
ChimeraX-ListInfo: 1.1.1
ChimeraX-Log: 1.1.5
ChimeraX-LookingGlass: 1.1
ChimeraX-Maestro: 1.8.2
ChimeraX-Map: 1.1.4
ChimeraX-MapData: 2.0
ChimeraX-MapEraser: 1.0.1
ChimeraX-MapFilter: 2.0.1
ChimeraX-MapFit: 2.0
ChimeraX-MapSeries: 2.1.1
ChimeraX-Markers: 1.0.1
ChimeraX-Mask: 1.0.2
ChimeraX-MatchMaker: 2.0.12
ChimeraX-MDcrds: 2.6
ChimeraX-MedicalToolbar: 1.0.2
ChimeraX-Meeting: 1.0.1
ChimeraX-MLP: 1.1.1
ChimeraX-mmCIF: 2.12
ChimeraX-MMTF: 2.2
ChimeraX-Modeller: 1.5.9
ChimeraX-ModelPanel: 1.3.7
ChimeraX-ModelSeries: 1.0.1
ChimeraX-Mol2: 2.0
ChimeraX-Mole: 1.0
ChimeraX-Morph: 1.0.2
ChimeraX-MouseModes: 1.2
ChimeraX-Movie: 1.0
ChimeraX-Neuron: 1.0
ChimeraX-Nifti: 1.0
ChimeraX-NRRD: 1.0
ChimeraX-Nucleotides: 2.0.3
ChimeraX-OpenCommand: 1.10.1
ChimeraX-PDB: 2.7.2
ChimeraX-PDBBio: 1.0
ChimeraX-PDBLibrary: 1.0.2
ChimeraX-PDBMatrices: 1.0
ChimeraX-PickBlobs: 1.0.1
ChimeraX-Positions: 1.0
ChimeraX-PresetMgr: 1.1
ChimeraX-PubChem: 2.1
ChimeraX-QScore: 1.0
ChimeraX-ReadPbonds: 1.0.1
ChimeraX-Registration: 1.1.1
ChimeraX-RemoteControl: 1.0
ChimeraX-RenderByAttr: 1.1
ChimeraX-RenumberResidues: 1.1
ChimeraX-ResidueFit: 1.0.1
ChimeraX-RestServer: 1.1
ChimeraX-RNALayout: 1.0
ChimeraX-RotamerLibMgr: 3.0
ChimeraX-RotamerLibsDunbrack: 2.0
ChimeraX-RotamerLibsDynameomics: 2.0
ChimeraX-RotamerLibsRichardson: 2.0
ChimeraX-SaveCommand: 1.5.1
ChimeraX-SchemeMgr: 1.0
ChimeraX-SDF: 2.0.1
ChimeraX-Segger: 1.0
ChimeraX-Segment: 1.0.1
ChimeraX-SelInspector: 1.0
ChimeraX-SeqView: 2.8.3
ChimeraX-Shape: 1.0.1
ChimeraX-Shell: 1.0.1
ChimeraX-Shortcuts: 1.1.1
ChimeraX-ShowSequences: 1.0.1
ChimeraX-SideView: 1.0.1
ChimeraX-Smiles: 2.1
ChimeraX-SmoothLines: 1.0
ChimeraX-SpaceNavigator: 1.0
ChimeraX-StdCommands: 1.10.3
ChimeraX-STL: 1.0.1
ChimeraX-Storm: 1.0
ChimeraX-StructMeasure: 1.1.2
ChimeraX-Struts: 1.0.1
ChimeraX-Surface: 1.0.1
ChimeraX-SwapAA: 2.0.1
ChimeraX-SwapRes: 2.2.1
ChimeraX-TapeMeasure: 1.0
ChimeraX-Test: 1.0
ChimeraX-Toolbar: 1.1.2
ChimeraX-ToolshedUtils: 1.2.1
ChimeraX-Topography: 1.0
ChimeraX-Tug: 1.0.1
ChimeraX-UI: 1.28.4
ChimeraX-uniprot: 2.2.2
ChimeraX-UnitCell: 1.0.1
ChimeraX-ViewDockX: 1.2
ChimeraX-VIPERdb: 1.0
ChimeraX-Vive: 1.1
ChimeraX-VolumeMenu: 1.0.1
ChimeraX-VTK: 1.0
ChimeraX-WavefrontOBJ: 1.0
ChimeraX-WebCam: 1.0.2
ChimeraX-WebServices: 1.1.1
ChimeraX-XMAS: 1.1.2
ChimeraX-Zone: 1.0.1
colorama: 0.4.6
comm: 0.1.3
contourpy: 1.0.7
cxservices: 1.2.2
cycler: 0.11.0
Cython: 0.29.33
debugpy: 1.6.7
decorator: 5.1.1
distro: 1.7.0
docutils: 0.19
et-xmlfile: 1.1.0
executing: 1.2.0
filelock: 3.9.0
fonttools: 4.39.3
funcparserlib: 1.0.1
grako: 3.16.5
h5py: 3.8.0
html2text: 2020.1.16
idna: 3.4
ihm: 0.35
imagecodecs: 2022.9.26
imagesize: 1.4.1
importlib-metadata: 6.6.0
ipykernel: 6.21.1
ipython: 8.10.0
ipython-genutils: 0.2.0
ipywidgets: 8.0.6
jedi: 0.18.2
Jinja2: 3.1.2
jupyter-client: 8.0.2
jupyter-core: 5.3.0
jupyterlab-widgets: 3.0.7
kiwisolver: 1.4.4
line-profiler: 4.0.2
lxml: 4.9.2
lz4: 4.3.2
MarkupSafe: 2.1.2
matplotlib: 3.6.3
matplotlib-inline: 0.1.6
msgpack: 1.0.4
nest-asyncio: 1.5.6
netCDF4: 1.6.2
networkx: 2.8.8
nibabel: 5.0.1
nptyping: 2.5.0
numexpr: 2.8.4
numpy: 1.23.5
openpyxl: 3.1.2
openvr: 1.23.701
packaging: 23.1
pandas: 2.0.3
ParmEd: 3.4.3
parso: 0.8.3
pep517: 0.13.0
pexpect: 4.8.0
pickleshare: 0.7.5
Pillow: 9.3.0
pip: 23.0
pkginfo: 1.9.6
platformdirs: 3.5.0
prompt-toolkit: 3.0.38
psutil: 5.9.4
ptyprocess: 0.7.0
pure-eval: 0.2.2
pycollada: 0.7.2
pydicom: 2.3.0
Pygments: 2.14.0
pynrrd: 1.0.0
PyOpenGL: 3.1.5
PyOpenGL-accelerate: 3.1.5
pyparsing: 3.0.9
pyproject-hooks: 1.0.0
PyQt6-commercial: 6.4.2
PyQt6-Qt6: 6.4.3
PyQt6-sip: 13.4.1
PyQt6-WebEngine-commercial: 6.4.0
PyQt6-WebEngine-Qt6: 6.4.3
python-dateutil: 2.8.2
pytz: 2023.3
pyzmq: 25.0.2
qtconsole: 5.4.0
QtPy: 2.3.1
QtRangeSlider: 0.1.5
RandomWords: 0.4.0
rdkit-pypi: 2022.9.2
requests: 2.28.2
scipy: 1.9.3
seaborn: 0.12.2
Send2Trash: 1.8.2
SEQCROW: 1.7.1
setuptools: 67.4.0
sfftk-rw: 0.7.3
six: 1.16.0
snowballstemmer: 2.2.0
sortedcontainers: 2.4.0
soupsieve: 2.4.1
sphinx: 6.1.3
sphinx-autodoc-typehints: 1.22
sphinxcontrib-applehelp: 1.0.4
sphinxcontrib-blockdiag: 3.0.0
sphinxcontrib-devhelp: 1.0.2
sphinxcontrib-htmlhelp: 2.0.1
sphinxcontrib-jsmath: 1.0.1
sphinxcontrib-qthelp: 1.0.3
sphinxcontrib-serializinghtml: 1.1.5
stack-data: 0.6.2
tables: 3.7.0
tcia-utils: 1.2.0
tifffile: 2022.10.10
tinyarray: 1.2.4
tomli: 2.0.1
tornado: 6.3.1
traitlets: 5.9.0
typing-extensions: 4.5.0
tzdata: 2023.3
urllib3: 1.26.15
wcwidth: 0.2.6
webcolors: 1.12
wheel: 0.38.4
wheel-filename: 1.4.1
widgetsnbextension: 4.0.7
zipp: 3.15.0
File attachment: v7n_from_chimerax_mol2.png
{kind=link}
{kind=link}
Attachments (6)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Change History (63)
by , 3 years ago
| Attachment: | v7n_from_chimerax_mol2.png added |
|---|
comment:1 by , 3 years ago
| Cc: | added |
|---|---|
| Component: | Unassigned → Structure Editing |
| Owner: | set to |
| Platform: | → all |
| Project: | → ChimeraX |
| Status: | new → accepted |
| Summary: | ChimeraX bug report submission → ESPALOMA-based parameter estimation |
| Type: | defect → task |
comment:4 by , 3 years ago
If my reading of section C.2.1 in the appendix is correct, Espaloma no longer requires bond orders and formal charges as input. Instead, the latest version uses the element type, hybridization, aromaticity, ring membership / sizes, mass, and number of bond partners for each atom -- all of which are already readily available in ChimeraX. So unless I'm mistaken, we "just" have to make Espaloma available in the ChimeraX ecosystem in order to use it for parameter/charge estimation.
comment:5 by , 3 years ago
If that’s correct, that’s awesome news! Last I’d heard from John (a few weeks ago), he was saying explicit bond orders were required - but he was excited at the prospect of trying with the approach I outlined. I’ve asked our software eng team if they might be able to help port it from conda. On Fri, 21 Jul 2023 at 19:04, ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote: > >
comment:6 by , 3 years ago
After poking around their GitHub (e.g. https://github.com/choderalab/espaloma/blob/main/espaloma/graphs/utils/read_homogeneous_graph.py) I'm pretty sure my reading is correct. Another challenge is somehow communicating that information directly from ChimeraX to Espaloma, rather than through an intermediate file where all that information is lost and needs to be recomputed.
comment:7 by , 3 years ago
I poked around the Espaloma GitHub repository some more, and am pretty sure that you can create an "espaloma model" without formal charges and bond orders, but am not clear whether you can convert to an "openMM system" without those (you have to have a molecular "heterograph", and it has charge / bond order info, but it's unclear if that info's needed for the conversion). So instead of poking around 3 GitHub repositories to try to determine these things (OpenMM, OpenFF, Espaloma), I think the next step is to install Conda so that I can interactively determine what are in these models and what the requirements actually are.
comment:8 by , 3 years ago
I am disappointed to report that for benzene, if you use single bonds everywhere then the charges are half the magnitude of what they should be. If you make the carbon-carbon bonds aromatic with order 1.5, then the charges are correct.
It might still be possible to do something here, but the trick would be to not mis-identify single bonds connecting aromatic rings as themselves aromatic -- particularly such bonds in non-aromatic ring systems. Also, double/triple bonds would need to be identified. So, some work -- basically #4270
comment:9 by , 3 years ago
I spoke slightly too soon. I get the correct charges with bond order 1, as long as the carbon bonds are marked as aromatic. I need to check out some systems that involve double bonds. If I _only_ have to mark bond aromaticity correctly, that is a significantly easier task than bond order also (given the info that ChimeraX has on hand [e.g. some conjugated bond systems would be rough to figure out]).
comment:10 by , 3 years ago
Something to keep in mind: if you do run into hard-to-surmount problems, we could always look into retraining based on the information ChimeraX *can* easily provide. Shouldn’t be hard for me to find the computational resources for that. On Wed, 30 Aug 2023 at 10:39, ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote: > >
comment:11 by , 3 years ago
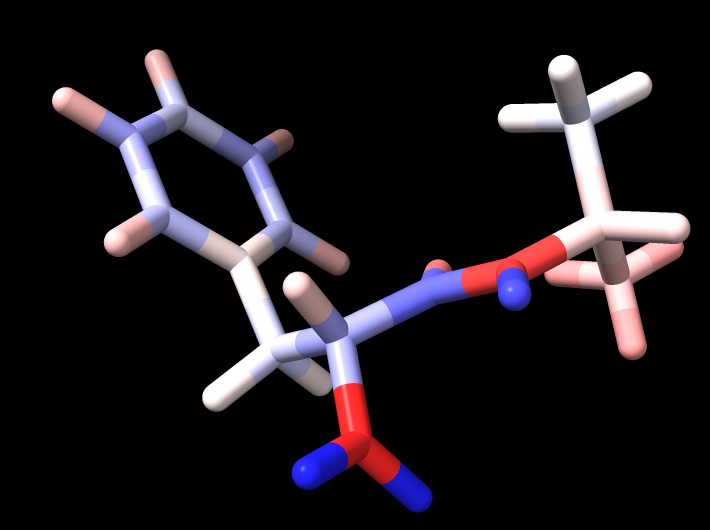
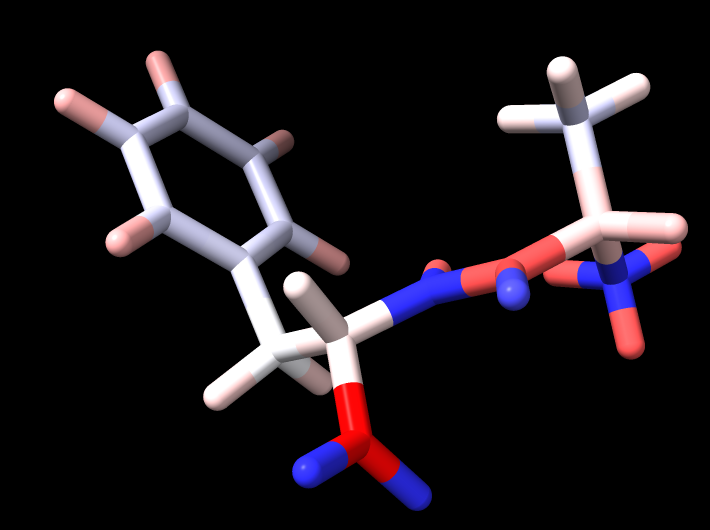
It seems that Espaloma and Amber charges differ markedly. For an alanine-phenylalanine dipeptide, if I only indicate the aromatic bonds and assign all bond orders as 1, then Espaloma charges differ from Amber charges by an average magnitude of .135 per atom. If I use bond order of 1.5 for the aromatic and resonance bonds, then the difference drops to .122 . The biggest difference is probably at the N terminus, where Amber assigns positive charge values to all the atoms, including the N, whereas Espaloma assigns a markedly negative charge to the N and high positive charges to the hydrogens.
I'm attaching 4 images. One is the dipeptide simply colored by element type. The second is colored by Amber charges. The third by (the less different) Espaloma charges. And the fourth by the difference in the two charge sets (Amber - Espaloma).
by , 3 years ago
| Attachment: | diff_true.png added |
|---|
Color by difference between Amber and Espaloma charges
comment:13 by , 3 years ago
Could see if John C. wants to join the thread to discuss the charge differences? On Sat, 2 Sep 2023 at 11:30, ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote: > >
comment:14 by , 3 years ago
Forgot to mention that I also had to specify formal charges (e.g. +1 for the N-terminal nitrogen).
I doubt getting John C. involved about the charge differences is worthwhile. I think Espaloma is well tested and we would use it if we can get it to work for us. The biggest difference (at the N-terminal ammonium cation) probably only makes a difference for short-range interactions where the higher positive charge on the hydrogens will make cation seem more positive than the Amber version. Since the combined N + hydrogens charge difference is minimal, at longer distances the cations will interact similarly.
I don't think I would want to use a system where I have to annotate the formal charges and bond orders accurately, so the question becomes how hard is it to train Espaloma just on IDATM types and bond connectivity. Would that be something we could do ourselves, or would John's group need to be involved?
comment:15 by , 3 years ago
Well… according to their paper and claims it’s *supposed* to be easy to retrain, but there’s always the question of how much that pans out in reality. We should be able to provide the computational resources… will make some enquiries once I’m back next week. On Fri, 15 Sep 2023 at 07:42, ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote: > > > >
comment:16 by , 3 years ago
So it looks like I should be able to get fairly easy access to a 8 x A100 instance. Will ask John for an estimate of how long retraining would take on that. On Thu, Sep 14, 2023 at 11:43 PM ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote: > > > >
comment:17 by , 3 years ago
Just got off a Zoom call with the bosses. They're keen to see this happen (as a separate, free open-source plugin that ISOLDE and any other MD-related plugins can rely on). Will discuss further with the local ML engineers next week. On Mon, Sep 25, 2023 at 12:01 PM ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote: > > > >
comment:18 by , 3 years ago
I have downloaded the SPICE dataset but haven't done anything with it yet. I was going to try to train, just learning partial charges, while screening out compounds that ChimeraX knows it isn't getting right (a warning about bond orders goes to stderr).
I guess we need to agree on how to proceed, who's doing what work, what the final product is, etc. Would you want other parameters learned besides partial charge? If it's packaged as a Toolshed plugin, how does ChimeraX itself use it in builtin code? (I can think of ways, but it's new ground for us). Since my work is theoretically owned by the regents of the university, there may be issues in making the code completely open source if I work on it.
I think we all want something that works well as quickly as feasible. Maybe this could be the plan(?):
- I produce a processed SPICE dataset that includes IDATM types and excludes structures that ChimeraX itself knows it's getting wrong.
- Your ML engineers use that dataset to train a network to produce whatever set a parameters we want to have.
- You make that into a Toolshed bundle.
Thoughts? The ChimeraX programmers would then have to figure out how distributed parts of ChimeraX can make use of functionality only available through the toolshed.
comment:19 by , 3 years ago
(Sorry for the slow reply - came down with a nasty virus over the weekend). To answer the (arguably) most important question up front: the catch to Altos providing resources to this is that (while they're happy to make it free open source) they do want to put some stamp of intellectual ownership on it - to be able to clearly point to it and say, "we did this" to earn some brownie points with the science community. I'm sure there's room for negotiation, but if the regents were to insist on making it non-free that would probably make things quite a bit more difficult. They've already made it clear they want it to be distinct from ISOLDE, since most of the benefits from advances to that still go to the University of Cambridge. So I guess the question is, what do you want? Do you want the extra work involved, or would you be OK with just letting us run with this one? Other questions: - Would you want other parameters learned besides partial charge? Yes, definitely. The ultimate aim here is to do away with the old-school AMBER residue-template approach to MD parameterisation and do as much as possible through ESPALOMA - i.e. use it to infer all the parameters necessary to run MD on a given model. Over time I'd then want to start exploring ways to extend its coverage to more exotic chemical space. - If it's packaged as a Toolshed plugin, how does ChimeraX itself use it in builtin code? In theory it wouldn't actually *have* to be a Toolshed bundle, since ultimately all it really needs from ChimeraX will be a graph representation of the model - which would only take a small amount of fairly straightforward code to make. So it could in principle just be put on the PyPI. Your strategy sounds fine (apart from the issues you've mentioned with ownership/licensing if you're actively involved). On Fri, Sep 29, 2023 at 9:30 PM ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote: > > > > > > > > >
comment:20 by , 3 years ago
Yeah, if my role is limited to providing a data file annotated with IDATM types and I'm not writing any actual code of the bundle/module, then I don't think it's actually an issue, though I'll run it by some people that are more lawyer-adjacent than me. :-)
comment:21 by , 3 years ago
I brought this up at our group meeting and it shouldn't be a problem. As an NIH grant recipient, any data we generate (such as an annotated/culled SPICE dataset) needs to be freely available.
comment:22 by , 3 years ago
Awesome! On Fri, Oct 6, 2023 at 2:28 AM ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote: > >
comment:23 by , 3 years ago
Just to give an update, I have a script that runs through the SPICE dataset and assigns IDATM types. Of the 19236 molecules, 5177 failed (26.9%), with the failures falling into these categories:
- 76 (1.5%) inorganic
- 38 (0.7%) could not determine bond orders in ring system
- 5063 (97.8%) protonation mismatch with atom types
I will be examining a sampling of the type 2 and 3 failures to see if I can convert some of those to successes. At least one low hanging fruit is that quite a few of the molecules have uncharged amino groups whereas ChimeraX makes them charged. I can just change the assignment from N3+ to N3 for those cases.
I also surveyed the atom types covered by the successful cases. The only "organic-y" types with no examples were O1, O1+, and C1-, which are tripled-bonded versions of those elements (there were 1819 examples of plain C1 though). The charged versions of those types are only found in carbon monoxide AFAIK. The minimum number of examples of any of the other atom types was 15.
comment:24 by , 3 years ago
So I decided to also screen out entries with multiple molecules, since those molecules are typically in close proximity and affect each other's charge distributions and sometimes protonation states, and our projected neural network will have no access to 3D proximity information. So of the original 19236 entries, I am screening out 3538 entries (18.4%; 33 inorganic, 3505 multiple molecules), leaving 15698 usable entries.
I did do the N3+ to N3 changeover as appropriate, making a dent in the protonation failures. There were 2073 total failures (13.1%) with:
- 37 (1.8%) could not determine bond orders in ring system
- 2036 (98.2%) protonation mismatch with atom types
The atom type coverage was much the same, with the lowest non-zero coverage dropping from 15 examples to 13.
I will continue investigating the failures as time permits.
comment:25 by , 3 years ago
So, Tristan, any idea what we should do if we find errors in the SPICE dataset? For instance the SPICE entry based on PubChem substance ID 386045997 has the proton on the 5-member ring on the wrong nitrogen, despite the protonation being correct in the corresponding PubChem compound entry (68785137).
comment:26 by , 3 years ago
(first, sorry for falling a bit silent on this - life has been unusually distracting lately). Will ask John Chodera for his advice - he’s the most likely contact I know. On Fri, 12 Jan 2024 at 18:29, ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote: > >
comment:27 by , 3 years ago
No problem. The ball's in my court currently anyway, though I'm getting closer to hitting it back over the net. :-)
comment:28 by , 3 years ago
Regarding your example, the 2D structure in the PubChem substance entry ( https://pubchem.ncbi.nlm.nih.gov/substance/386045997#section=2D-Structure&fullscreen=true) disagrees with the compound entry ( https://pubchem.ncbi.nlm.nih.gov/compound/68785137#section=2D-Structure) on which nitrogens in that ring are double-bonded. The former has the “lone” nitrogen single-bonded, whereas for the latter it’s one of the paired nitrogens. The behaviour of these triazoles isn’t my strongest suit, but looking at the PubChem entry (and Wikipedia page) for 1,2,4-triazole says that the compound entry is the correct one - which, as I understand it, is what you said. I take it the SPICE entry is protonated on the lone nitrogen? On Fri, 12 Jan 2024 at 18:36, ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote: > >
comment:29 by , 3 years ago
Geez, sorry to waste your time on this example. It is okay to have any one nitrogen protonated in that ring I think (though it's weird that it is protonated differently than the compound entry). The issue was that the bond lengths were throwing off the atom typing of the remaining nitrogens. However, yesterday I fixed a bug in my script about processing all the conformers (the bug resulting in a single conformer being examined multiple times) and one of the other conformers of this entry resulted in consistent atom typing -- so please go back to your other distractions for now. :-)
comment:30 by , 3 years ago
:-) Although I get the feeling that (absent any external influences) the lone nitrogen will have the lowest probability of being protonated. Probably someone’s done a (very dry) study on that somewhere. On Fri, 12 Jan 2024 at 19:29, ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote: > >
comment:31 by , 3 years ago
John suggests contacting Peter Eastman (peastman@stanford.edu) regarding SPICE issues, and says that since Peter is currently working on a SPICE 2.0 this would be a particularly useful time to do so. On Fri, Jan 12, 2024 at 7:39 PM ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote: > > > > >
comment:32 by , 3 years ago
Did some further work. Now screening out 3570 entries (18.6%; 33 inorganic, 3537 multiple molecules). Of the remaining 15666, there 1084 total failures (6.9%) with:
- 23 (2.1%) could not determine bond orders in ring system
- 940 (86.7%) protonation mismatch with atom types
- 121 (11.2%) not covered by IDATM typing (e.g. 5-bond phosphorus)
comment:33 by , 2 years ago
With the latest change, still screening out 3570 entries (18.6%), with 751 total failures (4.8%), of which:
1) 40 (5.3%) could not determine bond orders in ring system
2) 584 (77.8%) protonation mismatch with atom types
3) 127 (16.9%) not covered by IDATM typing (e.g. 5-bond phosphorus)
comment:34 by , 2 years ago
With more tweaks, still screening out 3570 entries (18.6%), with 742 total failures (4.7%), of which:
1) 37 (5.0%) could not determine bond orders in ring system
2) 578 (77.9%) protonation mismatch with atom types
3) 127 (17.1%) not covered by IDATM typing (e.g. 5-bond phosphorus)
comment:35 by , 2 years ago
Okay, the remaining failures seem to be largely for high valence states that have corresponding IDATM type, or disagreement on whether a triple-bonded carbon should be deprotonated and negatively charged or neutral and protonated. So I'm done with atom-type tweaking. The minimum number of examples of non O1/O1+/C1- types dropped from 15 to 14. Will have to think about what to do for the rare situations where those types show up.
So the next step is to rewrite a SPICE-like file containing just the usable molecules and their IDATM types.
comment:37 by , 2 years ago
| Status: | accepted → feedback |
|---|
I have a rewritten HDF5 file that has only the working entries, all their original info plus their IDATM types. It's 13.7 GB. How do you want me to get it to you? I could put it on plato, if you remember how to log into plato. I could put it at some URL fetchable from plato. What would you like?
comment:38 by , 2 years ago
This sounds like great progress! I've created a shared Google Drive folder and given you access (you should have received an email, but just in case the link is https://drive.google.com/drive/folders/1KUjahIo5nPtVTrpxukxZPOzj4PdCUm41?usp=sharing). Just drop it there?
A question regarding the 584 protonation-state mismatches: are these the sort of cases that might be worth leaving in anyway (i.e. tautomers that wouldn't be automatically assigned by AddH, but where the hydrogens could be manually added/removed by a savvy user later based on H-bonding, pH etc.)? Or do you think that would do more harm than good?
comment:39 by , 2 years ago
Even after "verifying my email" Google won't let me modify that folder in any way. Perhaps it would work better if you shared it with eric.pettersen@…?
As I mentioned, the protonation mismatches are largely high-valence states that don't have corresponding IDATM types, or protonated vs. negatively charged triple-bonded carbon (type C1/C1-). I only looked at a sampling of the failures, but not that many were because of tautomers. I also don't understand the motivation for leaving them in. You certainly don't want the massive mismatch between atoms and partial charges that would result.
comment:40 by , 2 years ago
Does that work now? The basis for me thinking it might be best to leave them in is that if they're present, then one would hope the algorithm *should* be able to learn to correctly assign charges (roughly, "this IDATM type in this context with a hydrogen gets charged this way, but without a hydrogen gets charged this other way")... but if they're left out, then they'll definitely be outside the training gamut so would likely yield nonsense if they turn up in a user's model. But I'm open to correction on that point. On Mon, Feb 26, 2024 at 10:28 PM ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote: > > >
comment:41 by , 2 years ago
That did work. The file is uploaded now.
The SPICE partial charges correspond to the SPICE protonation. If the protonation were different (e.g. the IDATM protonation) the partial charges on the atoms would be different. Those different charges are not in the file. How could the charges for the alternative protonation be used for learning when those charges are not in the data?
comment:42 by , 2 years ago
Probably just my misunderstanding, but what I was trying to get at is: what happens if / is it possible to keep the ChimeraX IDATM types on the heavy atoms, but force the protonation on those cases to match the SPICE topologies? I.e. to mimic the result of a user noticing that the default protonation doesn't match the environment, and manually removing/adding hydrogens to suit? Or is that just not sensible? On Mon, Feb 26, 2024 at 11:00 PM ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote: > > >
comment:43 by , 2 years ago
Atom types imply protonation. N3 is an sp3 nitrogen with 3 bonds. N3+ is an sp3 nitrogen with 4 bonds. ChimeraX will not assign atom types that indicate a proton should be removed, only possibly types that indicate that a proton should be added. For instance, if a structure has no hydrogens, a planar ring nitrogen might get assigned N2 (2 bonds, one double) or Npl (3 bonds, all single) or N2+ (3 bonds, one double, positively charged). But if a structure has hydrogens and that nitrogen has a proton, then the N2 atom type will never be assigned to it. But other ring nitrogens without explicit hydrogens might get assigned any of the three types including the 3-bond types, depending on what assignment ChimeraX thinks is best.
Maybe you mean change the IDATM types to match the SPICE protonation? I already do that to a limited extent, for instance changing N3+ to N3 when there are only 3 bonds, despite N3+ being the more likely at biological pH. Since most mismatches are for ring nitrogens and the minimum number of examples for those types in the current set is in the multiple hundreds, it would not seem to be worth the effort to do this.
I am concerned that there may not be enough examples of protonation at biological pH. I need to survey the dataset and see how many carboxylate moieties are protonated vs. unprotonated.
comment:44 by , 2 years ago
Since IDATM type Cac is only assigned for carboxylate carbons (C2 is used for carboxylic acid carbons), and there are 220 exemplars for Cac, I guess there are plenty of biological-pH-like molecules in the set.
comment:45 by , 2 years ago
Sorry it's been a while but we have made progress... but came across a real surprise in the code: for the ESPALOMA-SPICE experiment the code adding bond order information to the graph edges was commented out. Confirmed by looking at the details of the new ESPALOMA-SPICE preprint (https://arxiv.org/html/2307.07085v4/#A5): it seems that in my various conversations with John C. he wasn't fully aware of the latest details - they're no longer using explicit bond orders at all:
One of the improvements made from the previous Espaloma framework [121] is the exclusion of resonance-sensitive features, such as valences and formal charges, in order to improve the handling of molecules with atomic resonance, such as guanidinium and carboxylic acid. In this study, the input features of the atoms included the one-hot encoded element, as well as the hybridization, aromaticity, ring membership of sizes 3 to 8, atom mass, and the degree of the atoms, which is defined as the number of directly-bonded neighbors, all assigned using the RDKit 2023-03-4 release package [71].
That changes the equation pretty substantially, I think. All of the information they're applying to the graph should (I think?) now be trivially available directly from the ChimeraX API, so is there any actual reason left for retraining?
comment:46 by , 2 years ago
Unfortunately this sounds suspiciously like what I said in comment:4 of this ticket: "Instead, the latest version uses the element type, hybridization, aromaticity, ring membership / sizes, mass, and number of bond partners for each atom". Maybe it is actually true this time, but someone would have to revisit the tests done in comments 8, 9 and 11 to verify.
I mean, it would fantastic if it were true.
comment:47 by , 2 years ago
Oh, gods. My memory is a sieve sometimes (and I really should remember to skim back through threads before posting). Will do some more investigating.
by , 2 years ago
| Attachment: | lys_amide_charges.jpg added |
|---|
AM1BCC charges on lysine O-acetamide applied using "coulombic" command
comment:48 by , 2 years ago
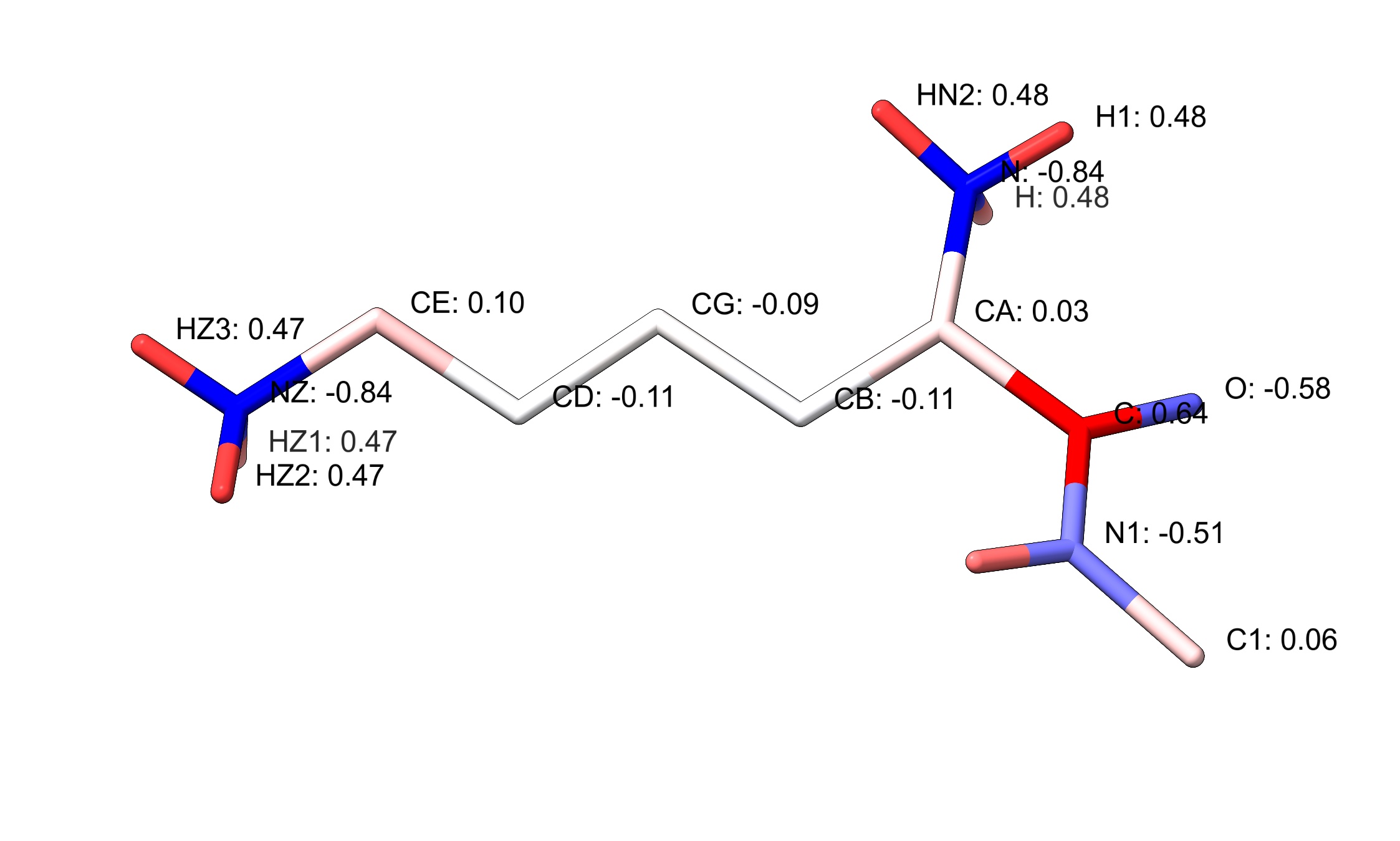
The differences in charges you observed look like they're just due to ESPALOMA using AM1BCC charges (actually, the OpenEye implementation am1bccelf10, but I don't think that's the important point) for everything, rather than the heavily hand-tweaked charges found in the core protein force field. See the attachment: if I add a C-terminal acetamide to a vanilla lysine residue and use the "coulombic" command to add charges to it, they come out looking very similar to what you describe in comment 11.
Seems to boil down to an inherent inconsistency between the AMBER protein and GAFF2 force fields (looking at the epsilon amine, the polarisation there is also much stronger - -0.84 vs -0.38 on the nitrogen; 0.47 vs 0.34 on each H). In theory one would hope that placing everything on a consistent charge framework (as opposed to the current scenario of slightly-to-markedly different for protein, nucleic acid, glycans and GAFF2 ligands) would lead to a better overall result... whether that plays out in reality remains to be seen.
comment:49 by , 2 years ago
An addition to the above: it turns out they're not actually training on the QM charges in the hdf5 dataset at all (I'm guessing probably because they're conformation dependent and would need some form of regularisation for equivalent atoms). It's all am1bccelf10 (https://docs.eyesopen.com/applications/quacpac/appendix.html). Not 100% sure of the wisdom of that approach, but that's how it stands.
comment:50 by , 2 years ago
Well I guess it does not matter if Espaloma charges differ from Amber charges, as long as the Espaloma charges produce accurate simulations in OpenMM, which it seems that they do. So is the upshot of all this that I should try to incorporate Espaloma directly into ChimeraX, or is your team going to continue to work on a bundle?
There is also the issue of how long does Espaloma take for structures of various sizes. If it's slow for whole proteins, we might want to continue with the current approach of only sending non-standard moieties into the computation and templating standard ones.
comment:51 by , 2 years ago
Second question first: attempting to run ESPALOMA on a 778-residue protein (AlphaFold-DB model for P15379, 11643 atoms after adding hydrogens) ran my 32GB system out of memory at the molecule_graph = esp.Graph(molecule) step (specifically, at https://github.com/choderalab/espaloma/blob/cb8e5b23e3ec1ada356128debc6a2a5511ef0b98/espaloma/graphs/utils/read_heterogeneous_graph.py#L271). So unless it can be made *drastically* more efficient (which would require understanding at a much deeper level than I currently have), it seems an all-at-once approach isn't yet on the cards.
In terms of implementation in ChimeraX: I'm open to discussion, but if (as it seems) there's currently no retraining needed then it probably makes most sense for you to incorporate it directly in ChimeraX.
comment:52 by , 2 years ago
Okay, I will investigate incorporating it directly and running it only on non-standard moieties.
comment:53 by , 2 years ago
We’ll still be looking into the potential for improvements, but not sure of the timeframe yet. On Thu, 30 May 2024 at 18:51, ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote: > >
comment:54 by , 2 years ago
So I tried using a smaller protein (IGF-I, 1010 atoms):
import espaloma as esp
from openff.toolkit import Topology
top = Topology.from_pdb('igf1_af2.pdb')
mol = top.molecule(0)
mgraph = esp.Graph(mol)
het = mgraph.heterograph
het.number_of_nodes()
1026779
het.number_of_edges()
6261550
That seems... a little excessive. I've asked a colleague who's a bit more familiar with graph ML approaches to help understand what's going on.
comment:55 by , 2 years ago
Did a little more testing with some smaller poly-A models... Nnodes ~ Natoms2 and Nedges ~ 6 * Natoms2. I'm thinking that has to be due to a mistake in the code... posted an issue on their GitHub: https://github.com/choderalab/espaloma/issues/217
comment:56 by , 2 years ago
If this proves to be a limitation instead of a bug, I am perfectly sanguine in continuing to use the current technique of extracting the non-standard moieties and parameterizing them separately. Problematic for something like a nanotube of course, but in practice mostly works fine.
comment:57 by , 10 months ago
| Resolution: | → fixed |
|---|---|
| Status: | feedback → closed |
Minimization has been implemented via Antechamber+OpenMM rather than with Espaloma. Would be trivial to implement dynamics if desired.
Added by email2trac