Opened 9 years ago
Closed 9 years ago
#804 closed defect (fixed)
Show residues fit error
| Reported by: | Owned by: | Tom Goddard | |
|---|---|---|---|
| Priority: | normal | Milestone: | |
| Component: | Volume Data | Version: | |
| Keywords: | Cc: | ||
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
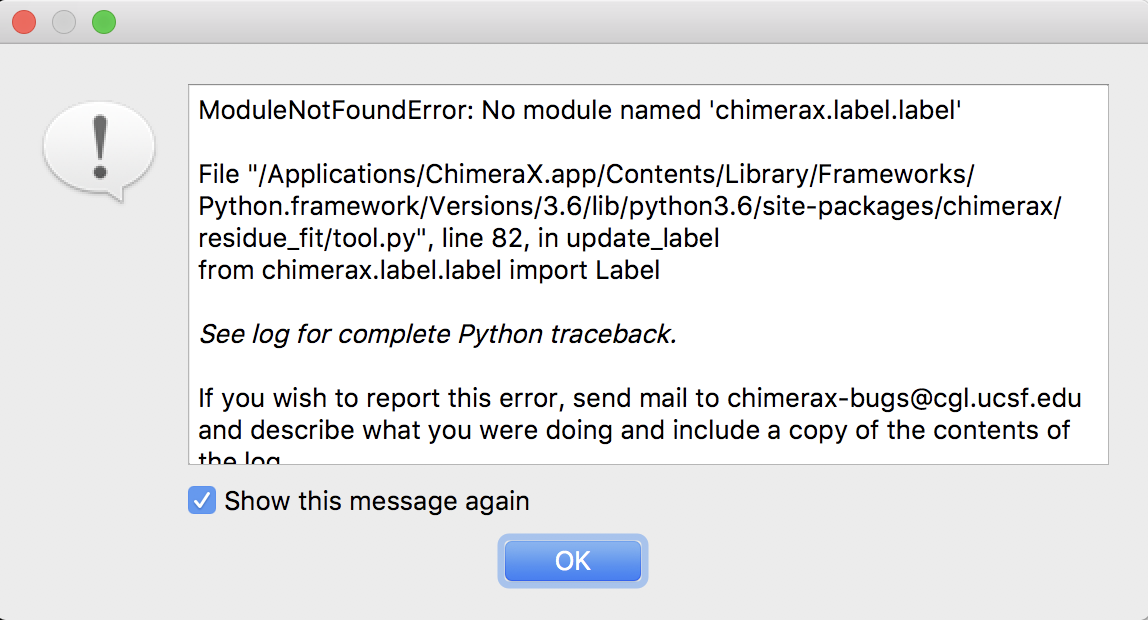
Hi, Tom, Congratulation on your new ChimeraX paper. We love ChimeraX with all its fancy new features. I just tried to show our protein residues fit in cryoEM density. With various current/recent version on MAC and PC, I keep getting the attached error message. However, older version 0.1-alpha1 didn't give me any error. Hope this information is helpful. Best wishes, Vinson Wen-Guang (Vinson) Liang, Ph.D Center for Integrative Science, Rm W423J (lab), Ben May Department for Cancer Research, The University of Chicago 929 E. 57th Street, Chicago, IL, 60637 Tel: 773-634-0798 (M) E-mail: <mailto:wliang@uchicago.edu> wliang@uchicago.edu; <mailto:wg.liang@hotmail.com> wg.liang@hotmail.com
{kind=link}
{kind=link}
Attachments (1)
Change History (3)
by , 9 years ago
| Attachment: | Chimerax_error.png added |
|---|
comment:1 by , 9 years ago
| Component: | Unassigned → Volume Data |
|---|---|
| Owner: | set to |
| Platform: | → all |
| Project: | → ChimeraX |
| Status: | new → assigned |
comment:2 by , 9 years ago
| Resolution: | → fixed |
|---|---|
| Status: | assigned → closed |
Fixed in tonight's ChimeraX builds.
Thanks for reporting this problem with the residue fit command. The label code that show the residue number in the graphics window had changed and that broke the residue fit code.
Note:
See TracTickets
for help on using tickets.
Added by email2trac