#7672 closed defect (fixed)
Delete and add label in same graphics update: could not broadcast input array
| Reported by: | Tristan Croll | Owned by: | Tom Goddard |
|---|---|---|---|
| Priority: | normal | Milestone: | |
| Component: | Depiction | Version: | |
| Keywords: | Cc: | Eric Pettersen | |
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
The following bug report has been submitted:
Platform: Windows-10-10.0.19044
ChimeraX Version: 1.4 (2022-06-03 23:39:42 UTC)
Description
Trying to remove a label and create new ones in the same graphics update fails.
Log:
UCSF ChimeraX version: 1.4 (2022-06-03)
© 2016-2022 Regents of the University of California. All rights reserved.
> open "C:/Users/Tristan
> Croll/Documents/Structures/trek-1_minispadin/6w8c_rebuild.cxs"
Opened (LIVE) 2mFo-DFc as #1.1.1.2, grid size 1,1,1, pixel 0.622,0.624,0.644,
shown at level 0.107, step 1, values float32
Opened (LIVE) mFo-DFc as #1.1.1.3, grid size 1,1,1, pixel 0.622,0.624,0.644,
shown at level -0.0678,0.0678, step 1, values float32
Opened (LIVE) 2mFo-DFc_sharp_12 as #1.1.1.4, grid size 1,1,1, pixel
0.622,0.624,0.644, shown at level 0.286, step 1, values float32
Opened (LIVE) MDFF potential as #1.1.1.5, grid size 1,1,1, pixel
0.622,0.624,0.644, shown at level 0.244, step 1, values float32
Restoring stepper: 6w8c
Log from Thu Sep 1 13:25:39 2022UCSF ChimeraX version: 1.4 (2022-06-03)
© 2016-2022 Regents of the University of California. All rights reserved.
> open "C:\\\Users\\\Tristan
> Croll\\\Documents\\\Structures\\\trek-1_minispadin\\\initial_dock.cxs"
Log from Fri Aug 26 09:33:11 2022UCSF ChimeraX version: 1.4 (2022-06-03)
© 2016-2022 Regents of the University of California. All rights reserved.
How to cite UCSF ChimeraX
> open 6w8c structureFactors true overSampling 2
Summary of feedback from opening 6w8c fetched from pdb
---
warning | WARNING: multiple experimental reflection datasets found:
F_meas_au, F_meas_sigma_au,
pdbx_F_plus, pdbx_F_plus_sigma, pdbx_F_minus, pdbx_F_minus_sigma,
pdbx_anom_difference, pdbx_anom_difference_sigma,
pdbx_I_plus, pdbx_I_plus_sigma, pdbx_I_minus, pdbx_I_minus_sigma,
pdbx_HL_A_iso, pdbx_HL_B_iso, pdbx_HL_C_iso, pdbx_HL_D_iso,
phase_calc, fom
Automatically choosing "F_meas_au, F_meas_sigma_au".
notes | Resolution: 2.600005818700715
Opened (LIVE) 2mFo-DFc as #1.1.1.2, grid size 42,40,38, pixel
0.622,0.624,0.644, shown at level 0.108, step 1, values float32
Opened (LIVE) mFo-DFc as #1.1.1.3, grid size 42,40,38, pixel
0.622,0.624,0.644, shown at level -0.0685,0.0685, step 1, values float32
Opened (LIVE) 2mFo-DFc_sharp_12 as #1.1.1.4, grid size 42,40,38, pixel
0.622,0.624,0.644, shown at level 0.289, step 1, values float32
Opened (STATIC) F_calc_au, phase_calc as #1.1.1.5, grid size 42,40,38, pixel
0.622,0.624,0.644, shown at level -0.233,0.233, step 1, values float32
Opened (STATIC) pdbx_DELFWT, pdbx_DELPHWT as #1.1.1.6, grid size 42,40,38,
pixel 0.622,0.624,0.644, shown at level -0.138,0.138, step 1, values float32
Opened (STATIC) pdbx_FWT, pdbx_PHWT as #1.1.1.7, grid size 42,40,38, pixel
0.622,0.624,0.644, shown at level -0.254,0.254, step 1, values float32
6w8c title:
K2P2.1 (TREK-1):ML335 complex, 1 mM K+ [more info...]
Chain information for 6w8c
---
Chain | Description | UniProt
1.2/A 1.2/B | Potassium channel subfamily K member 2 | KCNK2_MOUSE
Non-standard residues in 6w8c #1.2
---
11A — undecanoic acid
B7G — heptyl beta-D-glucopyranoside (HEPTYL-BETA-D-GLUCOPYRANOSIDE; heptyl
beta-D-glucoside; heptyl D-glucoside; heptyl glucoside)
CD — cadmium ion
D12 — dodecane
IEP —
[(2~{S})-1-octadecanoyloxy-3-[oxidanyl-[(1~{R},2~{R},3~{S},4~{S},5~{S},6~{S})-2,3,6-tris(oxidanyl)-4,5-diphosphonooxy-
cyclohexyl]oxy-phosphoryl]oxy-propan-2-yl] icosa-5,8,11,14-tetraenoate
K — potassium ion
LNK — pentane
OCT — N-octane
Q6F — N-[(2,4-dichlorophenyl)methyl]-4-[(methylsulfonyl)amino]benzamide
R16 — hexadecane
UND — undecane (lipid fragment)
> select ~prot
Expected an objects specifier or a keyword
> select ~protein
400 atoms, 381 bonds, 29 residues, 29 models selected
> clipper isolate sel
> delete ~protein&~:Q6F
> addh
Summary of feedback from adding hydrogens to 6w8c #1.2
---
warning | Not adding hydrogens to /B GLU 316 CB because it is missing heavy-
atom bond partners
notes | Termini for 6w8c (#1.2) chain A determined from SEQRES records
Termini for 6w8c (#1.2) chain B determined from SEQRES records
Chain-initial residues that are actual N termini:
Chain-initial residues that are not actual N termini: /A SER 35, /A SER 125,
/B SER 35, /B VAL 201
Chain-final residues that are actual C termini:
Chain-final residues that are not actual C termini: /A GLY 113, /A VAL 321, /B
TRP 199, /B GLU 316
531 hydrogen bonds
Adding 'H' to /A SER 35
Adding 'H' to /A SER 125
Adding 'H' to /B SER 35
Adding 'H' to /B VAL 201
/A VAL 321 is not terminus, removing H atom from 'C'
/B GLU 316 is not terminus, removing H atom from 'C'
4483 hydrogens added
> close #1.1
Deleting Crystallographic maps(6w8c-sf.cif)
Deleting (LIVE) 2mFo-DFc
Deleting (LIVE) mFo-DFc
Deleting (LIVE) 2mFo-DFc_sharp_12
> isolde ignore :Q6F
> set selectionWidth 4
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: Corrected atom nomenclature of 18 residues in model #1.2 to IUPAC-IUB
standards.
ISOLDE: currently ignoring 2 residues in model 1.2
Populating font family aliases took 189 ms. Replace uses of missing font
family "Carlito" with one that exists to avoid this cost.
> isolde restrain distances #1 kappa 40
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
> select clear
> set bgColor white
> ui tool show "Build Structure"
> build start peptide #1.2 GVSWGLR -139.0,135.0 -139.0,135.0 -139.0,135.0
> -139.0,135.0 -139.0,135.0 -139.0,135.0 -139.0,135.0 rotLib Dunbrack
> clipper spotlight
> addh
Summary of feedback from adding hydrogens to 6w8c #1.2
---
warning | Not adding hydrogens to /B GLU 316 CB because it is missing heavy-
atom bond partners
notes | Termini for 6w8c (#1.2) chain A determined from SEQRES records
Termini for 6w8c (#1.2) chain B determined from SEQRES records
No usable SEQRES records for 6w8c (#1.2) chain C; guessing termini instead
Chain-initial residues that are actual N termini: /C GLY 1, /C GLY 1
Chain-initial residues that are not actual N termini: /A SER 35, /A SER 125,
/B SER 35, /B VAL 201
Chain-final residues that are actual C termini: /C ARG 7, /C ARG 7
Chain-final residues that are not actual C termini: /A GLY 113, /A VAL 321, /B
TRP 199, /B GLU 316
502 hydrogen bonds
/A VAL 321 is not terminus, removing H atom from 'C'
/B GLU 316 is not terminus, removing H atom from 'C'
56 hydrogens added
> select /C
111 atoms, 112 bonds, 7 residues, 1 model selected
> ui mousemode right "translate selected atoms"
> select up
138 atoms, 138 bonds, 9 residues, 1 model selected
> select up
568 atoms, 574 bonds, 39 residues, 1 model selected
> isolde sim start sel
ISOLDE: started sim
> isolde sim pause
> isolde sim resume
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde sim stop
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: Corrected atom nomenclature of 4 residues in model #1.2 to IUPAC-IUB
standards.
ISOLDE: stopped sim
> delete /B:Q6F
> select up
24 atoms, 25 bonds, 1 residue, 1 model selected
> select up
111 atoms, 112 bonds, 7 residues, 1 model selected
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> cd /Users/tcroll/Documents/structures
Current working directory is: /Users/tcroll/Documents/structures
> cd /Users/tcroll/Documents/structures/trek-1_minispadin
Current working directory is:
/Users/tcroll/Documents/structures/trek-1_minispadin
> save initial_dock.cxs
——— End of log from Fri Aug 26 09:33:11 2022 ———
opened ChimeraX session
> isolde start
> set selectionWidth 4
> select up
111 atoms, 112 bonds, 7 residues, 1 model selected
> select up
8944 atoms, 9052 bonds, 565 residues, 1 model selected
> select down
111 atoms, 112 bonds, 7 residues, 1 model selected
> select up
138 atoms, 138 bonds, 9 residues, 1 model selected
> select up
568 atoms, 574 bonds, 39 residues, 1 model selected
> isolde sim start sel
ISOLDE: started sim
> select clear
> select up
24 atoms, 25 bonds, 1 residue, 1 model selected
> select up
111 atoms, 112 bonds, 7 residues, 1 model selected
> select zone sel 5
Selected 292 atoms
> select up
552 atoms, 544 bonds, 36 residues, 1 model selected
> isolde release distances #1.2&sel groupName "Reference Distance Restraints"
> select clear
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde sim stop
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> select up
11 atoms, 10 bonds, 1 residue, 1 model selected
> select up
111 atoms, 112 bonds, 7 residues, 1 model selected
> select zone sel 5 extend true
Selected 387 atoms
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop discardTo start
reverting to start
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> close #1
> open 6w8c structureFactors true overSampling 2.5
Summary of feedback from opening 6w8c fetched from pdb
---
warning | WARNING: multiple experimental reflection datasets found:
F_meas_au, F_meas_sigma_au,
pdbx_F_plus, pdbx_F_plus_sigma, pdbx_F_minus, pdbx_F_minus_sigma,
pdbx_anom_difference, pdbx_anom_difference_sigma,
pdbx_I_plus, pdbx_I_plus_sigma, pdbx_I_minus, pdbx_I_minus_sigma,
pdbx_HL_A_iso, pdbx_HL_B_iso, pdbx_HL_C_iso, pdbx_HL_D_iso,
phase_calc, fom
Automatically choosing "F_meas_au, F_meas_sigma_au".
notes | Fetching compressed mmCIF 6w8c from
http://files.rcsb.org/download/6w8c.cif
Fetching CCD K from http://ligand-expo.rcsb.org/reports/K/K/K.cif
Fetching CCD R16 from http://ligand-expo.rcsb.org/reports/R/R16/R16.cif
Fetching CCD Q6F from http://ligand-expo.rcsb.org/reports/Q/Q6F/Q6F.cif
Fetching CCD B7G from http://ligand-expo.rcsb.org/reports/B/B7G/B7G.cif
Fetching CCD IEP from http://ligand-expo.rcsb.org/reports/I/IEP/IEP.cif
Fetching CCD 11A from http://ligand-expo.rcsb.org/reports/1/11A/11A.cif
Fetching CCD CD from http://ligand-expo.rcsb.org/reports/C/CD/CD.cif
Fetching CCD OCT from http://ligand-expo.rcsb.org/reports/O/OCT/OCT.cif
Fetching CCD D12 from http://ligand-expo.rcsb.org/reports/D/D12/D12.cif
Fetching CCD UND from http://ligand-expo.rcsb.org/reports/U/UND/UND.cif
Fetching CCD LNK from http://ligand-expo.rcsb.org/reports/L/LNK/LNK.cif
Fetching compressed 6w8c structure factors from
http://files.rcsb.org/download/6w8c-sf.cif
Resolution: 2.6000058187007187
Opened (LIVE) 2mFo-DFc as #1.1.1.2, grid size 50,52,48, pixel
0.509,0.499,0.503, shown at level 0.107, step 1, values float32
Opened (LIVE) mFo-DFc as #1.1.1.3, grid size 50,52,48, pixel
0.509,0.499,0.503, shown at level -0.0678,0.0678, step 1, values float32
Opened (LIVE) 2mFo-DFc_sharp_12 as #1.1.1.4, grid size 50,52,48, pixel
0.509,0.499,0.503, shown at level 0.286, step 1, values float32
Opened (STATIC) F_calc_au, phase_calc as #1.1.1.5, grid size 50,52,48, pixel
0.509,0.499,0.503, shown at level -0.233,0.233, step 1, values float32
Opened (STATIC) pdbx_DELFWT, pdbx_DELPHWT as #1.1.1.6, grid size 50,52,48,
pixel 0.509,0.499,0.503, shown at level -0.138,0.138, step 1, values float32
Opened (STATIC) pdbx_FWT, pdbx_PHWT as #1.1.1.7, grid size 50,52,48, pixel
0.509,0.499,0.503, shown at level -0.255,0.255, step 1, values float32
6w8c title:
K2P2.1 (TREK-1):ML335 complex, 1 mM K+ [more info...]
Chain information for 6w8c
---
Chain | Description | UniProt
1.2/A 1.2/B | Potassium channel subfamily K member 2 | KCNK2_MOUSE
Non-standard residues in 6w8c #1.2
---
11A — undecanoic acid
B7G — heptyl beta-D-glucopyranoside (HEPTYL-BETA-D-GLUCOPYRANOSIDE; heptyl
beta-D-glucoside; heptyl D-glucoside; heptyl glucoside)
CD — cadmium ion
D12 — dodecane
IEP —
[(2~{S})-1-octadecanoyloxy-3-[oxidanyl-[(1~{R},2~{R},3~{S},4~{S},5~{S},6~{S})-2,3,6-tris(oxidanyl)-4,5-diphosphonooxy-
cyclohexyl]oxy-phosphoryl]oxy-propan-2-yl] icosa-5,8,11,14-tetraenoate
K — potassium ion
LNK — pentane
OCT — N-octane
Q6F — N-[(2,4-dichlorophenyl)methyl]-4-[(methylsulfonyl)amino]benzamide
R16 — hexadecane
UND — undecane (lipid fragment)
> close #1.1.1.5-7
> addh
Summary of feedback from adding hydrogens to 6w8c #1.2
---
warning | Not adding hydrogens to /B GLU 316 CB because it is missing heavy-
atom bond partners
notes | Termini for 6w8c (#1.2) chain A determined from SEQRES records
Termini for 6w8c (#1.2) chain B determined from SEQRES records
Chain-initial residues that are actual N termini:
Chain-initial residues that are not actual N termini: /A SER 35, /A SER 125,
/B SER 35, /B VAL 201
Chain-final residues that are actual C termini:
Chain-final residues that are not actual C termini: /A GLY 113, /A VAL 321, /B
TRP 199, /B GLU 316
548 hydrogen bonds
Adding 'H' to /A SER 35
Adding 'H' to /A SER 125
Adding 'H' to /B SER 35
Adding 'H' to /B VAL 201
/A VAL 321 is not terminus, removing H atom from 'C'
/B GLU 316 is not terminus, removing H atom from 'C'
5017 hydrogens added
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: Corrected atom nomenclature of 18 residues in model #1.2 to IUPAC-IUB
standards.
Opened (LIVE) MDFF potential as #1.1.1.5, grid size 50,52,48, pixel
0.509,0.499,0.503, shown at level 0.244, step 1, values float32
Loading residue template for 11A from internal database
Loading residue template for B7G from internal database
Loading residue template for D12 from internal database
Loading residue template for LNK from internal database
Loading residue template for OCT from internal database
Loading residue template for Q6F from internal database
Loading residue template for R16 from internal database
Loading residue template for UND from internal database
> delete sel
Traceback (most recent call last):
File "C:\Users\Tristan Croll\AppData\Local\UCSF\ChimeraX\1.4\site-
packages\chimerax\isolde\ui\validation_tab\unparameterised.py", line 160, in
_table_item_clicked_cb
residue.atoms.displays = True
File "atomic_cpp\cymol.pyx", line 994, in
chimerax.atomic.cymol.CyResidue.atoms.__get__
RuntimeError: Residue already deleted
RuntimeError: Residue already deleted
File "atomic_cpp\cymol.pyx", line 994, in
chimerax.atomic.cymol.CyResidue.atoms.__get__
See log for complete Python traceback.
> delete sel
> ui tool show "Ramachandran Plot"
QMainWindowLayout::tabPosition called with out-of-bounds value '0'
> select #1
9460 atoms, 9547 bonds, 34 pseudobonds, 584 residues, 30 models selected
> isolde sim start sel
ISOLDE: started sim
> select clear
> alphafold match #1
Fetching compressed AlphaFold P97438 from
https://alphafold.ebi.ac.uk/files/AF-P97438-F1-model_v3.cif
1 AlphaFold model found using UniProt identifier: P97438 (chains A,B)
AlphaFold chains matching 6w8c
---
Chain| UniProt Name| UniProt Id| RMSD| Length| Seen| % Id
A | KCNK2_MOUSE | P97438 | 1.93 | 302 | 276 | 96
B | KCNK2_MOUSE | P97438 | 1.97 | 302 | 281 | 96
Opened 2 AlphaFold models
> matchmaker #2.1 to #1.2
Parameters
---
Chain pairing | bb
Alignment algorithm | Needleman-Wunsch
Similarity matrix | BLOSUM-62
SS fraction | 0.3
Gap open (HH/SS/other) | 18/18/6
Gap extend | 1
SS matrix | | | H | S | O
---|---|---|---
H | 6 | -9 | -6
S | | 6 | -6
O | | | 4
Iteration cutoff | 2
Matchmaker 6w8c, chain A (#1.2) with AlphaFold KCNK2_MOUSE chain A, chain A
(#2.1), sequence alignment score = 1468.2
RMSD between 207 pruned atom pairs is 0.787 angstroms; (across all 276 pairs:
1.914)
> color #2.1 bychain
> color #2.1 byhetero
> color modify #2.1 hue + 50
ISOLDE: attempting to load PAE matrix for model #2.1 from the AlphaFold-EBI
database. If this fails or you wish to use a different PAE matrix, use the
"Load PAE matrix" button in ISOLDE's reference model restraints widget.
> alphafold pae #2.1 uniprotId P97438 plot false
Fetching compressed AlphaFold PAE P97438 from
https://alphafold.ebi.ac.uk/files/AF-P97438-F1-predicted_aligned_error_v3.json
Number of residues in structure "AlphaFold KCNK2_MOUSE chain A #2.1" is 302
which does not match PAE matrix size 426.
This can happen if the AlphaFold model has been trimmed to match an
experimental structure, or if residues have been deleted. The full-length
AlphaFold model must be used to show predicted aligned error.
> close #2
> alphafold match #1 trim false
1 AlphaFold model found using UniProt identifier: P97438 (chains A,B)
AlphaFold chains matching 6w8c
---
Chain| UniProt Name| UniProt Id| RMSD| Length| Seen| % Id
A | KCNK2_MOUSE | P97438 | 1.91 | 426 | 276 | 93
B | KCNK2_MOUSE | P97438 | 1.97 | 426 | 281 | 93
Opened 2 AlphaFold models
> matchmaker #2.1 to #1.2
Parameters
---
Chain pairing | bb
Alignment algorithm | Needleman-Wunsch
Similarity matrix | BLOSUM-62
SS fraction | 0.3
Gap open (HH/SS/other) | 18/18/6
Gap extend | 1
SS matrix | | | H | S | O
---|---|---|---
H | 6 | -9 | -6
S | | 6 | -6
O | | | 4
Iteration cutoff | 2
Matchmaker 6w8c, chain A (#1.2) with AlphaFold KCNK2_MOUSE chain A, chain A
(#2.1), sequence alignment score = 1468.2
RMSD between 207 pruned atom pairs is 0.788 angstroms; (across all 276 pairs:
1.915)
> color #2.1 bychain
> color #2.1 byhetero
> color modify #2.1 hue + 50
ISOLDE: attempting to load PAE matrix for model #2.1 from the AlphaFold-EBI
database. If this fails or you wish to use a different PAE matrix, use the
"Load PAE matrix" button in ISOLDE's reference model restraints widget.
> alphafold pae #2.1 uniprotId P97438 plot false
> isolde restrain torsions #1.2/A templateResidues #2.1/A adjustForConfidence
> true sidechains true springConstant 250.00 alpha 0.20
The "isolde restrain torsions" command only applies to protein chains. Other
residues have been ignored.
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: Corrected atom nomenclature of 4 residues in model #1.2 to IUPAC-IUB
standards.
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: Corrected atom nomenclature of 64 residues in model #2.1 to IUPAC-IUB
standards.
> isolde restrain torsions #1.2/B templateResidues #2.1/A adjustForConfidence
> true sidechains true springConstant 250.00 alpha 0.20
The "isolde restrain torsions" command only applies to protein chains. Other
residues have been ignored.
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: Corrected atom nomenclature of 4 residues in model #1.2 to IUPAC-IUB
standards.
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
> select clear
> hide #!2 models
> struts first
Missing or invalid "atoms" argument: invalid atoms specifier
> struts first
Missing or invalid "atoms" argument: invalid atoms specifier
> isolde shorthand
Initialising ISOLDE-specific command aliases:
Alias Equivalent full command
-------------------------------------------------
st isolde step {arguments}
aw isolde add water {arguments}
awsf isolde add water {arguments} sim false
al isolde add ligand {arguments}
aa isolde add aa $1 sel {arguments}
ht isolde mod his sel {arguments}
so setattr sel atoms occupancy {arguments}
ab isolde adjust bfactors {arguments}
ss isolde sim start sel
rt isolde release torsions sel {arguments}
rd isolde release distances sel {arguments}
ra rd; rt
pf isolde pepflip sel
cf isolde cisflip sel
cbb color bfactor {arguments}
cbo color byattr occupancy {arguments}
cbc color {arguments} bychain; color {arguments} byhet
cs clipper set contourSensitivity {arguments}
> st first
> ra
> select clear
> select up
12 atoms, 11 bonds, 1 residue, 1 model selected
> ra
> st
[Repeated 8 time(s)]
> select clear
> select up
22 atoms, 21 bonds, 1 residue, 1 model selected
> ra
> st
[Repeated 3 time(s)]
> ra
> st
[Repeated 17 time(s)]
> select up
12 atoms, 11 bonds, 1 residue, 1 model selected
> ra
> select clear
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: Corrected atom nomenclature of 6 residues in model #1.2 to IUPAC-IUB
standards.
ISOLDE: stopped sim
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> select :HOH
Nothing selected
> st
[Repeated 9 time(s)]
> isolde sim start sel
ISOLDE: started sim
> select clear
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
> delete sel
> select up
17 atoms, 17 bonds, 1 residue, 1 model selected
> ra
> isolde sim start sel
ISOLDE: started sim
> select clear
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 2 time(s)]
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 2 time(s)]
> isolde sim start sel
ISOLDE: started sim
> select clear
> ra
> select up
17 atoms, 16 bonds, 1 residue, 1 model selected
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
> select up
31 atoms, 29 bonds, 2 residues, 1 model selected
> select up
378 atoms, 378 bonds, 23 residues, 1 model selected
> isolde sim start sel
ISOLDE: started sim
> select clear
> show #!2 models
> hide #!2 models
> select up
7 atoms, 6 bonds, 1 residue, 1 model selected
> select up
28 atoms, 27 bonds, 3 residues, 1 model selected
> ra
> select up
14 atoms, 13 bonds, 1 residue, 1 model selected
> ra
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> select up
11 atoms, 10 bonds, 1 residue, 1 model selected
> ra
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> select up
17 atoms, 16 bonds, 1 residue, 1 model selected
> ra
> select up
19 atoms, 18 bonds, 1 residue, 1 model selected
> select up
78 atoms, 78 bonds, 5 residues, 1 model selected
> ra
> select clear
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> isolde sim start sel
ISOLDE: started sim
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 3 time(s)]
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 1 time(s)]
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 6 time(s)]
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 12 time(s)]
> show #!2 models
> hide #!2 models
> sequence chain #1/A
Alignment identifier is 1.2/A
> aa ILE
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 3 time(s)]
> show #!2 models
> hide #!2 models
> st
[Repeated 1 time(s)]
> ht both
Set protonation of HIS #1.2/A:126 to both
> st
[Repeated 7 time(s)]
> ra
> st
[Repeated 1 time(s)]
> aw
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 5 time(s)]
> delete sel
> st
[Repeated 4 time(s)]
> isolde sim start sel
ISOLDE: started sim
> select up
21 atoms, 20 bonds, 2 residues, 1 model selected
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 31 time(s)]
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: Corrected atom nomenclature of 1 residues in model #1.2 to IUPAC-IUB
standards.
ISOLDE: stopped sim
> st
[Repeated 5 time(s)]
> isolde sim start sel
ISOLDE: started sim
> ra
[Repeated 1 time(s)]
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: Corrected atom nomenclature of 1 residues in model #1.2 to IUPAC-IUB
standards.
ISOLDE: stopped sim
> st
[Repeated 7 time(s)]
> show #!2 models
> hide #!2 models
> select up
644 atoms, 649 bonds, 43 residues, 1 model selected
> select up
666 atoms, 670 bonds, 44 residues, 1 model selected
> select up
801 atoms, 809 bonds, 52 residues, 1 model selected
> isolde sim start sel
ISOLDE: started sim
> select clear
> show #!2 models
> show #2
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> hide #!2 models
> al CD
> select clear
> delete sel
> st
> al K
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop discardTo start
reverting to start
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> delete sel
> st
[Repeated 1 time(s)]
> show #!2 models
> hide #!2 models
> select up
157 atoms, 159 bonds, 9 residues, 1 model selected
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop discardTo start
reverting to start
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 6 time(s)]
> isolde sim start sel
ISOLDE: started sim
> select clear
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> select up
17 atoms, 16 bonds, 1 residue, 1 model selected
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 13 time(s)]
> ra
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> select clear
> st
[Repeated 3 time(s)]
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 1 time(s)]
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 11 time(s)]
> isolde sim start sel
ISOLDE: started sim
> ra
> cbb
16098 atoms, 1436 residues, atom bfactor range 29.9 to 276
> cbc
> ab 10
> select clear
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 19 time(s)]
> cbb
16098 atoms, 1436 residues, atom bfactor range 29.9 to 276
> cbc
> st
[Repeated 1 time(s)]
> aw
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 5 time(s)]
> select up
283 atoms, 286 bonds, 20 residues, 1 model selected
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: Corrected atom nomenclature of 1 residues in model #1.2 to IUPAC-IUB
standards.
ISOLDE: stopped sim
> st
[Repeated 2 time(s)]
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 2 time(s)]
> aw
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 20 time(s)]
> isolde sim start sel
ISOLDE: started sim
> select up
11 atoms, 10 bonds, 1 residue, 1 model selected
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 3 time(s)]
> ra
> st
[Repeated 1 time(s)]
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 3 time(s)]
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 4 time(s)]
> select up
832 atoms, 846 bonds, 51 residues, 1 model selected
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> al CD
> so 0
Assigning occupancy attribute to 1 item
> isolde sim start sel
ISOLDE: started sim
> select clear
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> delete sel
> st
[Repeated 3 time(s)]
> open 6w8c
6w8c title:
K2P2.1 (TREK-1):ML335 complex, 1 mM K+ [more info...]
Chain information for 6w8c #3
---
Chain | Description | UniProt
A B | Potassium channel subfamily K member 2 | KCNK2_MOUSE
Non-standard residues in 6w8c #3
---
11A — undecanoic acid
B7G — heptyl beta-D-glucopyranoside (HEPTYL-BETA-D-GLUCOPYRANOSIDE; heptyl
beta-D-glucoside; heptyl D-glucoside; heptyl glucoside)
CD — cadmium ion
D12 — dodecane
IEP —
[(2~{S})-1-octadecanoyloxy-3-[oxidanyl-[(1~{R},2~{R},3~{S},4~{S},5~{S},6~{S})-2,3,6-tris(oxidanyl)-4,5-diphosphonooxy-
cyclohexyl]oxy-phosphoryl]oxy-propan-2-yl] icosa-5,8,11,14-tetraenoate
K — potassium ion
LNK — pentane
OCT — N-octane
Q6F — N-[(2,4-dichlorophenyl)methyl]-4-[(methylsulfonyl)amino]benzamide
R16 — hexadecane
UND — undecane (lipid fragment)
> show #3
> close #3
> st
[Repeated 5 time(s)]
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 5 time(s)]
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #!2 models
> open 6w8c
6w8c title:
K2P2.1 (TREK-1):ML335 complex, 1 mM K+ [more info...]
Chain information for 6w8c #3
---
Chain | Description | UniProt
A B | Potassium channel subfamily K member 2 | KCNK2_MOUSE
Non-standard residues in 6w8c #3
---
11A — undecanoic acid
B7G — heptyl beta-D-glucopyranoside (HEPTYL-BETA-D-GLUCOPYRANOSIDE; heptyl
beta-D-glucoside; heptyl D-glucoside; heptyl glucoside)
CD — cadmium ion
D12 — dodecane
IEP —
[(2~{S})-1-octadecanoyloxy-3-[oxidanyl-[(1~{R},2~{R},3~{S},4~{S},5~{S},6~{S})-2,3,6-tris(oxidanyl)-4,5-diphosphonooxy-
cyclohexyl]oxy-phosphoryl]oxy-propan-2-yl] icosa-5,8,11,14-tetraenoate
K — potassium ion
LNK — pentane
OCT — N-octane
Q6F — N-[(2,4-dichlorophenyl)methyl]-4-[(methylsulfonyl)amino]benzamide
R16 — hexadecane
UND — undecane (lipid fragment)
> hide #!3 models
> isolde sim start sel
ISOLDE: started sim
> ra
> select clear
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 6 time(s)]
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 1 time(s)]
> isolde sim start sel
ISOLDE: started sim
> ra
> st
[Repeated 1 time(s)]
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 9 time(s)]
> isolde sim start sel
ISOLDE: started sim
> st
[Repeated 4 time(s)]
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 1 time(s)]
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 6 time(s)]
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 12 time(s)]
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: Corrected atom nomenclature of 1 residues in model #1.2 to IUPAC-IUB
standards.
ISOLDE: stopped sim
> st
[Repeated 6 time(s)]
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> select clear
> st
[Repeated 6 time(s)]
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 15 time(s)]
> ab 20
> select clear
> st
[Repeated 3 time(s)]
> select up
24 atoms, 22 bonds, 2 residues, 1 model selected
> select up
150 atoms, 152 bonds, 11 residues, 1 model selected
> isolde sim start sel
ISOLDE: started sim
> select clear
[Repeated 1 time(s)]
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> select clear
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> select clear
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: Corrected atom nomenclature of 1 residues in model #1.2 to IUPAC-IUB
standards.
ISOLDE: stopped sim
> st
[Repeated 10 time(s)]
> isolde sim start sel
ISOLDE: started sim
> select clear
> ab 20
[Repeated 1 time(s)]
> select clear
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 6 time(s)]
> ra
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 15 time(s)]
> isolde sim start sel
ISOLDE: started sim
> show #!3 models
> show #3
> hide #!3 models
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 27 time(s)]
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: Corrected atom nomenclature of 1 residues in model #1.2 to IUPAC-IUB
standards.
ISOLDE: stopped sim
> st
[Repeated 5 time(s)]
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 1 time(s)]
> show #!2 models
> hide #!2 models
> st
[Repeated 4 time(s)]
> select up
618 atoms, 623 bonds, 41 residues, 1 model selected
> isolde sim start sel
ISOLDE: started sim
> show #!2 models
> hide #!2 models
> show #!2 models
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> sequence chain #1/B
Alignment identifier is 1.2/B
> hide #!2 models
> aa ASN
> isolde sim start sel
ISOLDE: started sim
> select up
24 atoms, 25 bonds, 1 residue, 1 model selected
> ra
> isolde pepflip sel
Flipping the peptide bond for 1 residues
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 11 time(s)]
> isolde sim start sel
ISOLDE: started sim
> select up
17 atoms, 16 bonds, 1 residue, 1 model selected
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 12 time(s)]
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 3 time(s)]
> select up
38 atoms, 37 bonds, 1 residue, 1 model selected
> delete sel
> st
[Repeated 34 time(s)]
> aw
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 6 time(s)]
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 5 time(s)]
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: Corrected atom nomenclature of 1 residues in model #1.2 to IUPAC-IUB
standards.
ISOLDE: stopped sim
> st
[Repeated 5 time(s)]
> select up
34 atoms, 33 bonds, 1 residue, 1 model selected
> delete sel
> st
[Repeated 14 time(s)]
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
> isolde sim start sel
ISOLDE: started sim
> select up
24 atoms, 25 bonds, 1 residue, 1 model selected
> ra
[Repeated 1 time(s)]
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> select up
26 atoms, 25 bonds, 1 residue, 1 model selected
> select down
1 atom, 1 residue, 1 model selected
> select up
26 atoms, 25 bonds, 1 residue, 1 model selected
> delete sel
> isolde sim start sel
ISOLDE: started sim
> select clear
[Repeated 1 time(s)]
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 5 time(s)]
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> st
[Repeated 11 time(s)]
> ra
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> ra
> st
[Repeated 5 time(s)]
> swapaa mousemode sel GLU
> isolde sim start sel
ISOLDE: started sim
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> isolde sim start sel
ISOLDE: started sim
> select clear
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> sequence chain #1/B
Alignment identifier is 1.2/B
> aa TRP
> isolde sim start sel
ISOLDE: started sim
> ra
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> select up
38 atoms, 37 bonds, 1 residue, 1 model selected
> delete sel
> select up
26 atoms, 25 bonds, 1 residue, 1 model selected
> delete sel
[Repeated 1 time(s)]
> select clear
> select #1
9370 atoms, 9464 bonds, 33 pseudobonds, 583 residues, 35 models selected
> isolde sim start sel
ISOLDE: started sim
> select clear
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> select up
17 atoms, 16 bonds, 1 residue, 1 model selected
> delete sel
> select ~protein
892 atoms, 859 bonds, 51 residues, 35 models selected
> clipper isolate sel maskRadius 4.0 focus false
> select clear
> select up
50 atoms, 49 bonds, 1 residue, 1 model selected
> delete sel
> select up
100 atoms, 98 bonds, 2 residues, 1 model selected
> delete sel
> select up
34 atoms, 33 bonds, 1 residue, 1 model selected
> delete sel
> select up
45 atoms, 45 bonds, 1 residue, 1 model selected
> delete sel
> select up
45 atoms, 45 bonds, 1 residue, 1 model selected
> delete sel
> select up
45 atoms, 45 bonds, 1 residue, 1 model selected
> delete sel
> select up
45 atoms, 45 bonds, 1 residue, 1 model selected
> delete sel
> clipper spotlight
> select #1
8989 atoms, 9088 bonds, 33 pseudobonds, 574 residues, 35 models selected
> isolde sim start sel
ISOLDE: started sim
> select clear
> isolde sim stop
Updating bulk solvent parameters...
ISOLDE: Checking and correcting nomenclature for (pseudo)symmetric side
chains...
ISOLDE: stopped sim
> pwd
Current working directory is: C:\windows\system32
> cd "C:/Users/Tristan Croll/Documents/Structures/trek-1_minispadin"
Current working directory is: C:\Users\Tristan
Croll\Documents\Structures\trek-1_minispadin
> save 6w8c_rebuild.cxs
Taking snapshot of stepper: 6w8c
——— End of log from Thu Sep 1 13:25:39 2022 ———
opened ChimeraX session
> open 6w8c
6w8c title:
K2P2.1 (TREK-1):ML335 complex, 1 mM K+ [more info...]
Chain information for 6w8c #4
---
Chain | Description | UniProt
A B | Potassium channel subfamily K member 2 | KCNK2_MOUSE
Non-standard residues in 6w8c #4
---
11A — undecanoic acid
B7G — heptyl beta-D-glucopyranoside (HEPTYL-BETA-D-GLUCOPYRANOSIDE; heptyl
beta-D-glucoside; heptyl D-glucoside; heptyl glucoside)
CD — cadmium ion
D12 — dodecane
IEP —
[(2~{S})-1-octadecanoyloxy-3-[oxidanyl-[(1~{R},2~{R},3~{S},4~{S},5~{S},6~{S})-2,3,6-tris(oxidanyl)-4,5-diphosphonooxy-
cyclohexyl]oxy-phosphoryl]oxy-propan-2-yl] icosa-5,8,11,14-tetraenoate
K — potassium ion
LNK — pentane
OCT — N-octane
Q6F — N-[(2,4-dichlorophenyl)methyl]-4-[(methylsulfonyl)amino]benzamide
R16 — hexadecane
UND — undecane (lipid fragment)
> view #4/A:Q6F
> hide #!4 models
> close #2-4
> select clear
> ui tool show "Build Structure"
> build start peptide "custom built" GVSWGLR -139.0,135.0 -139.0,135.0
> -139.0,135.0 -139.0,135.0 -139.0,135.0 -139.0,135.0 -139.0,135.0 rotLib
> Dunbrack
Chain information for custom built #2
---
Chain | Description
A | No description available
> select #2
55 atoms, 56 bonds, 7 residues, 1 model selected
> ui mousemode right "translate selected atoms"
> addh #2
Summary of feedback from adding hydrogens to custom built #2
---
notes | No usable SEQRES records for custom built (#2) chain A; guessing
termini instead
Chain-initial residues that are actual N termini: custom built #2/A GLY 1
Chain-initial residues that are not actual N termini:
Chain-final residues that are actual C termini: custom built #2/A ARG 7
Chain-final residues that are not actual C termini:
0 hydrogen bonds
56 hydrogens added
> hide HC
> setattr #2 atoms occupancy 0
Assigning occupancy attribute to 111 items
> hide #2 models
> select up
37 atoms, 38 bonds, 1 residue, 1 model selected
> label sel residues
> ~label
> select clear
> select up
37 atoms, 38 bonds, 1 residue, 1 model selected
> label sel residues text "ML335 agonist "
> select clear
> select up
37 atoms, 38 bonds, 1 residue, 1 model selected
> color sel lightgreen
> color sel byhetero
> rock y 40 120 cycle 120
> rock y 40 240 cycle 120
> movie record
> rock y 40 240 cycle 120
> movie crossfade 20
> ~label
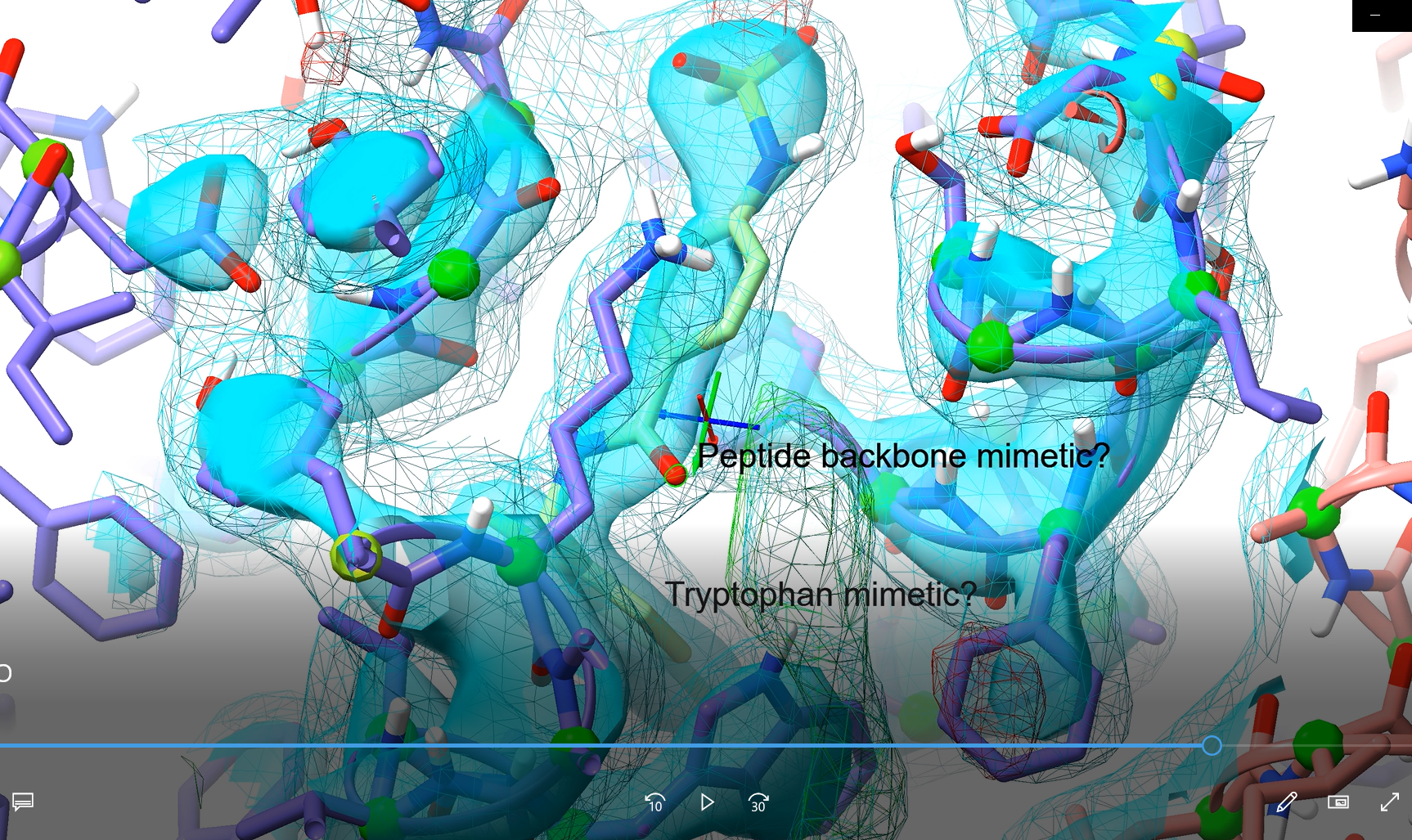
> label /A:408@C19 atoms text "Tryptophan mimetic? "; label /A:408@C13 atom
> text "peptide backbone? "
Traceback (most recent call last):
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\core\triggerset.py", line 134, in invoke
return self._func(self._name, data)
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\label\label3d.py", line 503, in _update_graphics_if_needed
self._rebuild_label_graphics()
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\label\label3d.py", line 513, in _rebuild_label_graphics
trgba, tcoord = self._packed_texture() # Compute images first since vertices
depend on image size
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\label\label3d.py", line 570, in _packed_texture
trgba[y:y+h,x:x+w,:] = rgba
ValueError: could not broadcast input array from shape (72,2090,4) into shape
(72,2048,4)
Error processing trigger "graphics update":
ValueError: could not broadcast input array from shape (72,2090,4) into shape
(72,2048,4)
File "C:\Program Files\ChimeraX\bin\lib\site-
packages\chimerax\label\label3d.py", line 570, in _packed_texture
trgba[y:y+h,x:x+w,:] = rgba
See log for complete Python traceback.
OpenGL version: 3.3.0 NVIDIA 472.98
OpenGL renderer: NVIDIA GeForce RTX 3070 Laptop GPU/PCIe/SSE2
OpenGL vendor: NVIDIA Corporation
Python: 3.9.11
Locale: en_GB.cp1252
Qt version: PyQt6 6.3.0, Qt 6.3.0
Qt runtime version: 6.3.0
Qt platform: windows
Manufacturer: HP
Model: HP ZBook Studio 15.6 inch G8 Mobile Workstation PC
OS: Microsoft Windows 10 Pro (Build 19044)
Memory: 34,007,068,672
MaxProcessMemory: 137,438,953,344
CPU: 16 11th Gen Intel(R) Core(TM) i7-11800H @ 2.30GHz
OSLanguage: en-GB
Installed Packages:
alabaster: 0.7.12
appdirs: 1.4.4
Babel: 2.10.1
backcall: 0.2.0
blockdiag: 3.0.0
certifi: 2022.5.18.1
cftime: 1.6.0
charset-normalizer: 2.0.12
ChimeraX-AddCharge: 1.2.3
ChimeraX-AddH: 2.1.11
ChimeraX-AlignmentAlgorithms: 2.0
ChimeraX-AlignmentHdrs: 3.2.1
ChimeraX-AlignmentMatrices: 2.0
ChimeraX-Alignments: 2.4.3
ChimeraX-AlphaFold: 1.0
ChimeraX-AltlocExplorer: 1.0.2
ChimeraX-AmberInfo: 1.0
ChimeraX-Arrays: 1.0
ChimeraX-Atomic: 1.39.1
ChimeraX-AtomicLibrary: 7.0
ChimeraX-AtomSearch: 2.0.1
ChimeraX-AxesPlanes: 2.1
ChimeraX-BasicActions: 1.1
ChimeraX-BILD: 1.0
ChimeraX-BlastProtein: 2.1.1
ChimeraX-BondRot: 2.0
ChimeraX-BugReporter: 1.0
ChimeraX-BuildStructure: 2.7
ChimeraX-Bumps: 1.0
ChimeraX-BundleBuilder: 1.1
ChimeraX-ButtonPanel: 1.0
ChimeraX-CageBuilder: 1.0
ChimeraX-CellPack: 1.0
ChimeraX-Centroids: 1.2
ChimeraX-ChemGroup: 2.0
ChimeraX-Clashes: 2.2.4
ChimeraX-Clipper: 0.19.0.dev0
ChimeraX-ColorActions: 1.0
ChimeraX-ColorGlobe: 1.0
ChimeraX-ColorKey: 1.5.1
ChimeraX-CommandLine: 1.2.3
ChimeraX-ConnectStructure: 2.0.1
ChimeraX-Contacts: 1.0
ChimeraX-Core: 1.4
ChimeraX-CoreFormats: 1.1
ChimeraX-coulombic: 1.3.2
ChimeraX-Crosslinks: 1.0
ChimeraX-Crystal: 1.0
ChimeraX-CrystalContacts: 1.0
ChimeraX-DataFormats: 1.2.2
ChimeraX-Dicom: 1.1
ChimeraX-DistMonitor: 1.1.5
ChimeraX-Dssp: 2.0
ChimeraX-EMDB-SFF: 1.0
ChimeraX-ExperimentalCommands: 1.0
ChimeraX-FileHistory: 1.0
ChimeraX-FunctionKey: 1.0
ChimeraX-Geometry: 1.2
ChimeraX-gltf: 1.0
ChimeraX-Graphics: 1.1
ChimeraX-Hbonds: 2.1.2
ChimeraX-Help: 1.2
ChimeraX-HKCage: 1.3
ChimeraX-IHM: 1.1
ChimeraX-ImageFormats: 1.2
ChimeraX-IMOD: 1.0
ChimeraX-IO: 1.0.1
ChimeraX-ISOLDE: 1.4
ChimeraX-ItemsInspection: 1.0
ChimeraX-Label: 1.1.1
ChimeraX-ListInfo: 1.1.1
ChimeraX-Log: 1.1.5
ChimeraX-LookingGlass: 1.1
ChimeraX-Maestro: 1.8.1
ChimeraX-Map: 1.1
ChimeraX-MapData: 2.0
ChimeraX-MapEraser: 1.0
ChimeraX-MapFilter: 2.0
ChimeraX-MapFit: 2.0
ChimeraX-MapSeries: 2.1
ChimeraX-Markers: 1.0
ChimeraX-Mask: 1.0
ChimeraX-MatchMaker: 2.0.6
ChimeraX-MDcrds: 2.6
ChimeraX-MedicalToolbar: 1.0.1
ChimeraX-Meeting: 1.0
ChimeraX-MLP: 1.1
ChimeraX-mmCIF: 2.7
ChimeraX-MMTF: 2.1
ChimeraX-Modeller: 1.5.5
ChimeraX-ModelPanel: 1.3.2
ChimeraX-ModelSeries: 1.0
ChimeraX-Mol2: 2.0
ChimeraX-Morph: 1.0
ChimeraX-MouseModes: 1.1
ChimeraX-Movie: 1.0
ChimeraX-Neuron: 1.0
ChimeraX-Nucleotides: 2.0.2
ChimeraX-OpenCommand: 1.9
ChimeraX-PDB: 2.6.6
ChimeraX-PDBBio: 1.0
ChimeraX-PDBLibrary: 1.0.2
ChimeraX-PDBMatrices: 1.0
ChimeraX-PickBlobs: 1.0
ChimeraX-Positions: 1.0
ChimeraX-PresetMgr: 1.1
ChimeraX-PubChem: 2.1
ChimeraX-ReadPbonds: 1.0.1
ChimeraX-Registration: 1.1
ChimeraX-RemoteControl: 1.0
ChimeraX-ResidueFit: 1.0
ChimeraX-RestServer: 1.1
ChimeraX-RNALayout: 1.0
ChimeraX-RotamerLibMgr: 2.0.1
ChimeraX-RotamerLibsDunbrack: 2.0
ChimeraX-RotamerLibsDynameomics: 2.0
ChimeraX-RotamerLibsRichardson: 2.0
ChimeraX-SaveCommand: 1.5.1
ChimeraX-SchemeMgr: 1.0
ChimeraX-SDF: 2.0
ChimeraX-Segger: 1.0
ChimeraX-Segment: 1.0
ChimeraX-SelInspector: 1.0
ChimeraX-SeqView: 2.6
ChimeraX-Shape: 1.0.1
ChimeraX-Shell: 1.0
ChimeraX-Shortcuts: 1.1
ChimeraX-ShowAttr: 1.0
ChimeraX-ShowSequences: 1.0
ChimeraX-SideView: 1.0
ChimeraX-Smiles: 2.1
ChimeraX-SmoothLines: 1.0
ChimeraX-SpaceNavigator: 1.0
ChimeraX-StdCommands: 1.8
ChimeraX-STL: 1.0
ChimeraX-Storm: 1.0
ChimeraX-StructMeasure: 1.0.1
ChimeraX-Struts: 1.0.1
ChimeraX-Surface: 1.0
ChimeraX-SwapAA: 2.0
ChimeraX-SwapRes: 2.1.1
ChimeraX-TapeMeasure: 1.0
ChimeraX-Test: 1.0
ChimeraX-Toolbar: 1.1.1
ChimeraX-ToolshedUtils: 1.2.1
ChimeraX-Tug: 1.0
ChimeraX-UI: 1.18.3
ChimeraX-uniprot: 2.2
ChimeraX-UnitCell: 1.0
ChimeraX-ViewDockX: 1.1.2
ChimeraX-VIPERdb: 1.0
ChimeraX-Vive: 1.1
ChimeraX-VolumeMenu: 1.0
ChimeraX-VTK: 1.0
ChimeraX-WavefrontOBJ: 1.0
ChimeraX-WebCam: 1.0
ChimeraX-WebServices: 1.1.0
ChimeraX-Zone: 1.0
colorama: 0.4.4
comtypes: 1.1.10
cxservices: 1.2
cycler: 0.11.0
Cython: 0.29.26
debugpy: 1.6.0
decorator: 5.1.1
docutils: 0.17.1
entrypoints: 0.4
filelock: 3.4.2
fonttools: 4.33.3
funcparserlib: 1.0.0
grako: 3.16.5
h5py: 3.7.0
html2text: 2020.1.16
idna: 3.3
ihm: 0.27
imagecodecs: 2021.11.20
imagesize: 1.3.0
ipykernel: 6.6.1
ipython: 7.31.1
ipython-genutils: 0.2.0
jedi: 0.18.1
Jinja2: 3.0.3
jupyter-client: 7.1.0
jupyter-core: 4.10.0
kiwisolver: 1.4.2
line-profiler: 3.4.0
lxml: 4.7.1
lz4: 3.1.10
MarkupSafe: 2.1.1
matplotlib: 3.5.1
matplotlib-inline: 0.1.3
msgpack: 1.0.3
nest-asyncio: 1.5.5
netCDF4: 1.5.8
networkx: 2.6.3
numexpr: 2.8.1
numpy: 1.22.1
openvr: 1.16.802
packaging: 21.3
ParmEd: 3.4.3
parso: 0.8.3
pickleshare: 0.7.5
Pillow: 9.0.1
pip: 21.3.1
pkginfo: 1.8.2
prompt-toolkit: 3.0.29
psutil: 5.9.0
pycollada: 0.7.2
pydicom: 2.2.2
Pygments: 2.11.2
PyOpenGL: 3.1.5
PyOpenGL-accelerate: 3.1.5
pyparsing: 3.0.9
PyQt6-commercial: 6.3.0
PyQt6-Qt6: 6.3.0
PyQt6-sip: 13.3.1
PyQt6-WebEngine-commercial: 6.3.0
PyQt6-WebEngine-Qt6: 6.3.0
python-dateutil: 2.8.2
pytz: 2022.1
pywin32: 303
pyzmq: 23.1.0
qtconsole: 5.3.0
QtPy: 2.1.0
RandomWords: 0.3.0
requests: 2.27.1
scipy: 1.7.3
setuptools: 59.8.0
sfftk-rw: 0.7.2
six: 1.16.0
snowballstemmer: 2.2.0
sortedcontainers: 2.4.0
Sphinx: 4.3.2
sphinx-autodoc-typehints: 1.15.2
sphinxcontrib-applehelp: 1.0.2
sphinxcontrib-blockdiag: 3.0.0
sphinxcontrib-devhelp: 1.0.2
sphinxcontrib-htmlhelp: 2.0.0
sphinxcontrib-jsmath: 1.0.1
sphinxcontrib-qthelp: 1.0.3
sphinxcontrib-serializinghtml: 1.1.5
suds-community: 1.0.0
tables: 3.7.0
tifffile: 2021.11.2
tinyarray: 1.2.4
tornado: 6.1
traitlets: 5.1.1
urllib3: 1.26.9
wcwidth: 0.2.5
webcolors: 1.11.1
wheel: 0.37.1
wheel-filename: 1.3.0
WMI: 1.5.1
Attachments (1)
{kind=link}
Change History (7)
comment:1 by , 4 years ago
| Cc: | added |
|---|---|
| Component: | Unassigned → Depiction |
| Owner: | set to |
| Platform: | → all |
| Project: | → ChimeraX |
| Status: | new → assigned |
| Summary: | ChimeraX bug report submission → Delete and add label in same graphics update: could not broadcast input array |
comment:2 by , 4 years ago
| Resolution: | → fixed |
|---|---|
| Status: | assigned → closed |
follow-up: 3 comment:3 by , 4 years ago
That’s odd - I haven’t done anything to the default label size, and they printed fine when I entered the commands separately. The display I was using was on the higher-resolution size, but not crazily so. On Mon, 26 Sep 2022 at 19:58, ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote:
comment:4 by , 4 years ago
The error message indicates one of the two labels had pixel size 2090 wide by 72 high. The text "Tryptophan mimetic? " is 20 characters and the pixel aspect ratio is 29:1, seems unexpectedly wide. That label comes out as 446 by 54 on my MacBook Pro with retina display. Qt is asked to provide the font at a default 48 point font and who knows what it used on your Windows machine. Then Qt makes the image from the font.
I was able to reproduce the bug using a much longer label text and specifying larger size than the default 48.
label sel atoms text "Tryptophan mimetic? And maybe something more" size 100 ValueError: could not broadcast input array from shape (112,2198,4) into shape (112,2048,4)
follow-up: 5 comment:5 by , 4 years ago
This is what it looked like when I repeated the same sequence of commands one-by-one (still from the resulting movie; movie dimensions are 2970x1770). Overkill really - I didn't realise that display was quite that high-res. [image: image.png] On Mon, Sep 26, 2022 at 11:40 PM ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote:

comment:6 by , 4 years ago
When I said the label texture was more than 2048 pixels wide this does not mean it would appear that wide on the screen. It is just the texture size which will be scaled when rendered onto the screen.
I think we are missing something. But I easily reproduced your error using long label text and am pretty sure that is the underlying cause.
Fixed.
The error is because the label width exceeds the width of the texture (2048 pixels). I guess you have set the default label size to something quite large. Ideally the code would increase the texture size, but it is not able to do that currently. I've increased the label texture width to 4096 and put in code that clips bigger labels and issues a warning that the label was too wide and was clipped (rightmost part of text not shown). A single texture packs all the labels to allow efficient rendering. It could be improved to reallocate the texture to larger sizes (although many OpenGL implementations will limit it to maximum width 16384). But since this error has never been reported before I think it is not currently worth the time and complexity to implement the dynamics resizing of the texture.