Opened 9 years ago
Closed 8 years ago
#694 closed enhancement (fixed)
Add C-alpha trace depictions
| Reported by: | Owned by: | Tom Goddard | |
|---|---|---|---|
| Priority: | major | Milestone: | |
| Component: | Depiction | Version: | |
| Keywords: | Cc: | tic20@…, conrad@…, meng@…, pett@… | |
| Blocked By: | Blocking: | ||
| Notify when closed: | v.kalienkova@bioc.uzh.ch | Platform: | all |
| Project: | ChimeraX |
Description
Begin forwarded message:
From: Oliver Clarke
Subject: [chimerax-users] Suggestion - add shortcuts for Ca trace, Ca+sidechains
Date: May 26, 2017 at 7:35:46 AM PDT
To: chimerax-users@…
Hi,
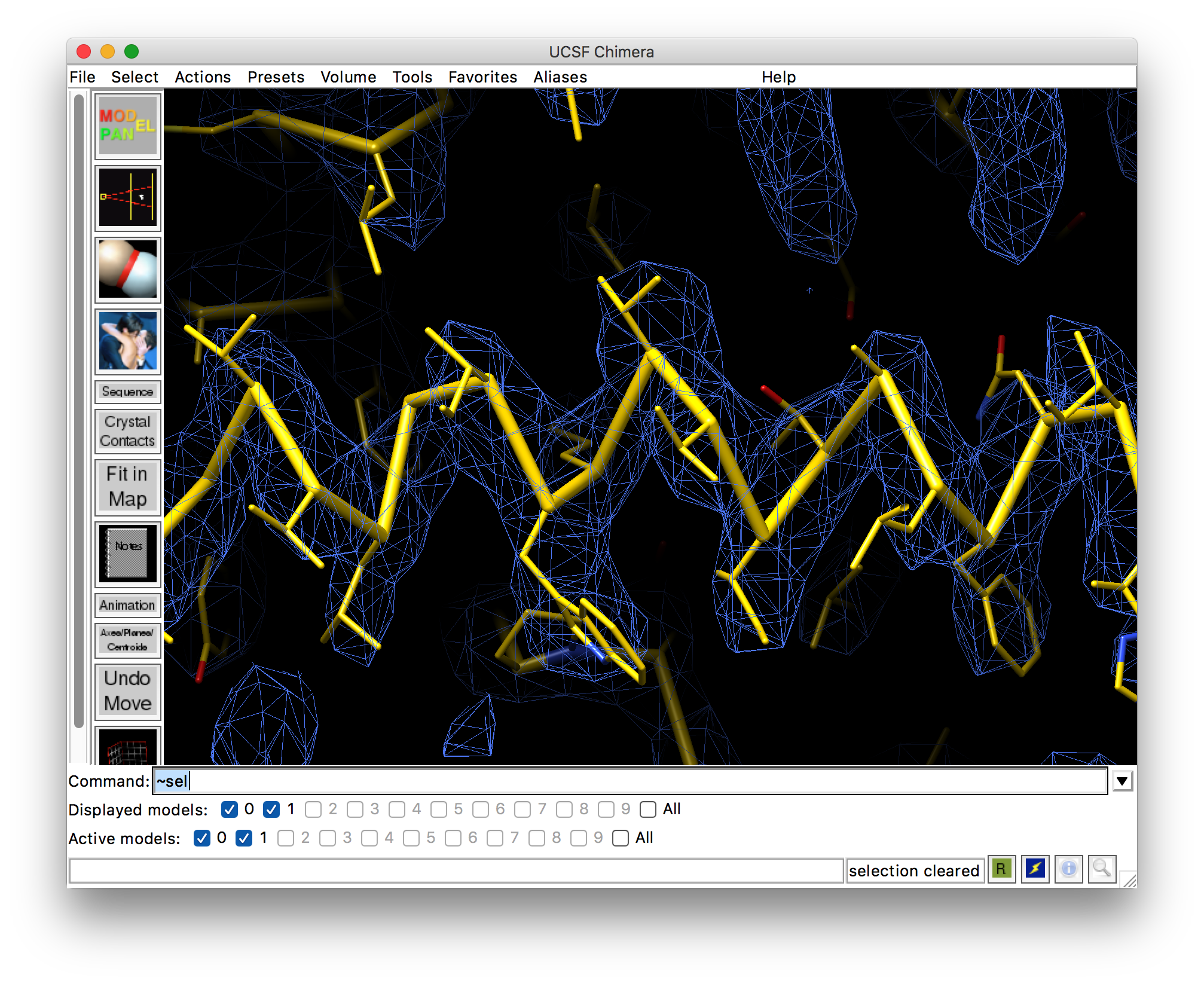
Frequently when looking at models in medium resolution maps (~4-5 Å), it is valuable to view the C-alpha trace when assessing model map fit (or making figures to show such).
At slightly higher resolution, where side chains are obvious but backbone torsions cannot be modeled based on the map alone, representing the C-alpha trace with sidechains displayed is often desirable (examples of both attached).
It looks like there is space in the upper toolbar for additional buttons - perhaps adding a button for a Calpha trace representation (equivalent would be P for nuclei acids) and one for a Calpha+sidechain/base representation may be worth considering? (I’m also not convinced of the value of the random atom colors representation as a shortcut, but maybe there is a use case I’m missing?)
Cheers
Oli
Attachments (2)
{kind=link}
{kind=link}
{kind=link}
Change History (33)
by , 9 years ago
| Attachment: | Screenshot 2017-05-26 10.34.38.png added |
|---|
comment:1 by , 9 years ago
comment:2 by , 9 years ago
Hmm - not too fussed but there are definitely instances where having a direct c-alpha trace - not ribbons - is helpful. Having the ribbon is nice, but the user doesn’t know where the Calphas are unless you explicitly point it out to them Oli
follow-up: 2 comment:3 by , 8 years ago
| Cc: | added |
|---|
comment:4 by , 8 years ago
| Cc: | removed |
|---|---|
| Owner: | changed from to |
Tristan Croll also requests C-alpha chain traces with straight segments between CA atoms.
Reassigning to Eric to figure out the state in atomic data structures for handling chain trace display.
Begin forwarded message:
From: Tristan Croll
Subject: Re: Timeline for ChimeraX 0.7?
Date: July 24, 2018 at 10:21:04 AM PDT
To: Eric Pettersen
Hi Eric,
The visualisation Isabel (and Airlie, and quite a few other experimental folks, for that matter) is after is much simpler - just direct, straight bonds between CAs. It has advantages over the spline when building - namely, you can see instantly exactly where the CA is, and where the CB should go.
As I said, it’s easy enough for me to mock up a workable solution using pseudobonds, so no real pressure from me. But worth noting that there is interest from the community in having the option available.
Cheers,
Tristan
comment:5 by , 8 years ago
| Cc: | conrad@cgl.ucsf.edu, meng@cgl.ucsf.edu, tic20@cam.ac.uk → tic20@cam.ac.uk, conrad@cgl.ucsf.edu, meng@cgl.ucsf.edu |
|---|
comment:6 by , 8 years ago
| Notify when closed: | → v.kalienkova@bioc.uzh.ch |
|---|
comment:7 by , 8 years ago
Would a boolean 'chain_trace' (or 'draw_trace'?) Chain attribute be sufficient? In conjunction with the existing 'polymer_type attribute, I think that would be enough to draw a chain trace. That would make chain tracing controllable on a per-chain basis rather than per model.
comment:8 by , 8 years ago
| Cc: | added |
|---|---|
| Status: | assigned → accepted |
comment:9 by , 8 years ago
Sounds good! So long as it is possible to set as a global preference Cheers Oli
follow-up: 9 comment:10 by , 8 years ago
A per-chain "chain_trace" attribute could work. Would it be true by default? What would the precise rules be for when the CA-CA dashed lines are shown? Maybe Elaine can weigh in on that. Would it just auto-show when only the CA of a residue is displayed, or the ribbon with no atoms shown?
comment:11 by , 8 years ago
I should think that chain_trace==true means show the backbone as simple _solid_ lines -- no ribbons, no non-CA/P backbone atoms. So, obviously it would default false.
I guess that setting chain_trace to true would have to result in the ribbon_display of each Residue of the Chain to false, and conversely setting the ribbon_display of any Residue of the Chain to true would set the Chain's chain_trace to false.
comment:12 by , 8 years ago
Or, alternatively chain_trace==true could simple "override" ribbon drawing in the graphics code and not touch the ribbon_display attribute. The question then would be when does chain tracing get turned off -- explicitly by a command (~chain) or implicitly by backbone atom or ribbon display. I guess Elaine would be the best one to determine what would be most natural.
comment:13 by , 8 years ago
I thought chain_trace = true meant something different than both your ideas. I thought it did not effect ribbon or atom display in any way. It just means that when only the CA is the only atom shown for two adjacent residues then a dashed line is draw between them. I guess it would be useful to hear from Oliver and Tristan if the chain trace should show a ball for the CA atoms or only the lines with no atom indicated.
follow-up: 14 comment:15 by , 8 years ago
Keep in mind ChimeraX does not show wires, so the lines are cylinders and so there will be at least a ball of the same radius as the cylinder to join two cylinders.
follow-up: 15 comment:16 by , 8 years ago
| Cc: | added; removed |
|---|---|
| Owner: | changed from to |
| Status: | accepted → assigned |
So the upshot of an offline discussion Tom and I had is that chaining should work the same as when Chimera1's auto-chaining is on. So there would be no explicit attribute that tracked chain tracing (AtomicStructures would always chain trace when appropriate [i.e. showing CAs only] and Structures would not).
Re-assigning to T.G.
comment:17 by , 8 years ago
I would generally agree that it makes sense to have the chain trace automatically appear when the CAs are the only *backbone* atoms shown (I can imagine situations where CA trace + sidechains would be appropriate). I think solid rather than dashed bonds would be better, though. Tristan Croll Research Fellow Cambridge Institute for Medical Research University of Cambridge CB2 0XY
follow-up: 17 comment:18 by , 8 years ago
It is definitely useful to be able to show CAs and sidechains - I do this all the time for low res structures, it is much easier to interpret. Agreed, solid cylinders not dashed lines. Cheers Oli
follow-up: 18 comment:19 by , 8 years ago
I added auto chain tracing for proteins and nucleic acids. If consecutive CA atoms in a chain are shown and the C and N atoms between two CA atoms are hidden or do not exist then a chain trace pseudobond is shown between the CA atoms. For nucleic acids consecutive P atoms are used and O3' and O5' must be undisplayed.
There is a Structure attribute "autochain" that determines whether autochaining is done. The default will be true. But this is currently disabled in the code until ChimeraX 0.8 is released (possibly tomorrow). It is disabled by having autochain false -- if you wish to try it before the 0.8 release use command "setattr #!1 m autochain true" for model #1.
If autochain pseudobonds are shown they belong to a submodel of the structure called "chain trace". That submodel disappears if atom display changes such that no chain trace is shown.
The pseudobonds are cylinders, not dashed, are halfbond colored, and are half the radius of the stick atoms. I know Oliver asked that the atoms not be visible which could be done by making the pseudobonds have radius equal to the stick atom radius, but I think you will be happier that I used half that radius because otherwise they are pretty fat. This default behavior can be changed if we decide it is not optimal.
comment:20 by , 8 years ago
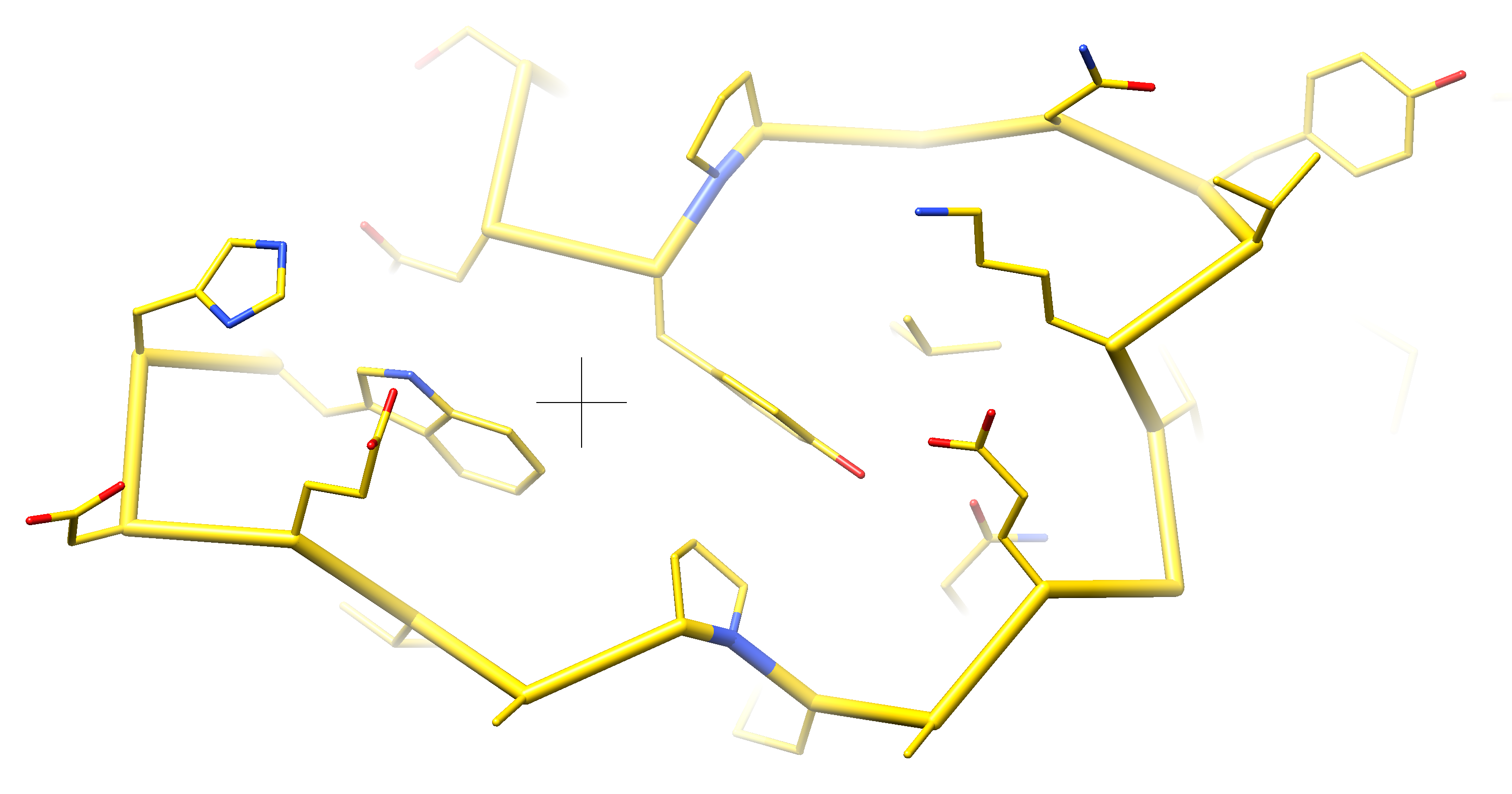
This sounds good! The one query/request I would have is to make an exception for prolines, such that if CAs and sidechains are shown, the N of the proline is included in the trace, so that the sidechain of proline is not all broken and weird looking. This is handled correctly (IMO) in chimera (see attached):

follow-up: 20 comment:21 by , 8 years ago
I would suggest the tweak of having autochaining initialized as false for Structures and true for AtomicStructures.
comment:22 by , 8 years ago
The simple chain trace use which I am guessing is 99% of cases is only CA are shown, not side chains. So I am not going to spend the time to make prolines (and nucleic acid sugars) handle chain traces that connect other backbone atoms. I'm willing to here arguments that in fact chain trace with side chains shown is used all the time. If it is rare and you want it all the same, we should make another ticket. The current chain trace behavior will not show a chain trace bond from the proline to the preceding residue if the proline N is shown.
comment:23 by , 8 years ago
Well I can only speak for myself, but I use it all the time for looking at models (and making figures) in low resolution (3-5Å) single particle EM maps. I would imagine this is a significant use case for ChimeraX but maybe not. I use CA+sidechains representation at least as often as CA-only, likely more. I don’t think nucleic acid sugars need this (they are a constant feature of the backbone). The only exception I can think of is proline. Cheers Oli
comment:24 by , 8 years ago
It turns out that CA-only structures show dashed line "missing structure" segments instead of a chain trace. The chain trace between two CA currently requires that a real bond connects those two residues, so chain trace is not shown in this case. Eric suggests leaving this behavior as is. He says that "missing structure" includes any case where adjacent residues do not have the expected polymer bond between them. (It does not mean a residue is missing, as I thought.) Eric says this is important because connectivity is used for defining chains and those missing structure pseudobonds establish the connectivity in CA-only structures.
follow-up: 23 comment:25 by , 8 years ago
This I (personally) care much less about - CA-only structures are pretty rare these days. Even at low res I would suggest best practice is to deposit poly-ALA (or poly-UNK), for precisely this reason - visualization/depiction of CA-only structures is a mess. Cheers Oli
comment:26 by , 8 years ago
I only mention the CA-only case for clarification. I think it behaves weirdly treating those as "missing structure" and not chain tracing them and simply wanted to document why it works that way.
follow-up: 25 comment:27 by , 8 years ago
I made a separate ticket #1528 for improving chain trace for prolines with side chains shown.
comment:28 by , 8 years ago
Still need to change autochain to default true. Currently (version 0.9 2018-12-22) it is false by default. Example:
open 1bzm
~cartoon
hide
show @ca | sidechain
setattr struct autochain true
...can then use "size" command to fatten the trace to the same as the sticks, or conversely, make the real-bond sticks thinner.
comment:29 by , 8 years ago
Oh, I see Eric's previous comment:
I would suggest the tweak of having autochaining initialized as false for
Structures and true for AtomicStructures.
Not sure how to distinguish those two in the "setattr" command, but perhaps if it is already true for atomic structures, one would not need to use that command to adjust autochaining.
comment:30 by , 8 years ago
Okay, I implemented my own suggestion of defaulting chain-tracing to 'on' for AtomicStructures. Can be reverted or tweaked if it proves problematic.
comment:31 by , 8 years ago
| Resolution: | → fixed |
|---|---|
| Status: | assigned → closed |
Chain trace display for atomic structures is on by default now.
We don't current display lines between C-alpha atoms when only the C-alphas are shown, nor do we have a command that can do that.
Eric and Elaine and I discussed the options.
One option that can be used in current ChimeraX is to show a ribbon tube that goes through the C-alpha atoms, for instance with commands
cartoon style #1 arrows f wid .3 thick .3 x round
cartoon #1 smooth 0
Two other options that are not implemented in ChimeraX but are in Chimera 1 are to automatically create pseudobonds, or have a command ("chain") that creates those pseudobonds. The automatic creation and destruction is quite complex. The command would require also issuing a command to hide the CA trace pseudobonds.
Another option is to improve the ribbon capabilities so it can draw straight tubes between the C-alpha atoms.