Opened 4 years ago
Last modified 4 years ago
#6590 assigned enhancement
AlphaFold PAE domains should exclude disordered regions
| Reported by: | Owned by: | Tom Goddard | |
|---|---|---|---|
| Priority: | normal | Milestone: | |
| Component: | Structure Prediction | Version: | |
| Keywords: | Cc: | Tristan Croll | |
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
The following bug report has been submitted:
Platform: macOS-12.3.1-arm64-arm-64bit
ChimeraX Version: 1.4.dev202203271748 (2022-03-27 17:48:18 UTC)
Description
AlphaFold PAE domain coloring often groups long disordered regions (100 disordered residues or more) with regions that have secondary structure. It would be nice if those long disordered regions were not part of real domains. One idea for doing this would be to not put residues in domains if their pLDDT score is below a some threshold (e.g. 70).
Log:
UCSF ChimeraX version: 1.4.dev202203271748 (2022-03-27)
© 2016-2022 Regents of the University of California. All rights reserved.
How to cite UCSF ChimeraX
> alphafold fetch BCR_HUMAN
Chain information for AlphaFold BCR_HUMAN #1
---
Chain | Description | UniProt
A | Breakpoint cluster region protein | BCR_HUMAN
> alphafold pae #1 uniprotId BCR_HUMAN colorDomains true
OpenGL version: 4.1 Metal - 76.3
OpenGL renderer: Apple M1 Max
OpenGL vendor: Apple
Locale: UTF-8
Qt version: PyQt6 6.2.3, Qt 6.2.3
Qt platform: cocoa
Hardware:
Hardware Overview:
Model Name: MacBook Pro
Model Identifier: MacBookPro18,2
Chip: Apple M1 Max
Total Number of Cores: 10 (8 performance and 2 efficiency)
Memory: 32 GB
System Firmware Version: 7459.101.3
OS Loader Version: 7459.101.3
Software:
System Software Overview:
System Version: macOS 12.3.1 (21E258)
Kernel Version: Darwin 21.4.0
Time since boot: 6 days 8:32
Graphics/Displays:
Apple M1 Max:
Chipset Model: Apple M1 Max
Type: GPU
Bus: Built-In
Total Number of Cores: 32
Vendor: Apple (0x106b)
Metal Family: Supported, Metal GPUFamily Apple 7
Displays:
Color LCD:
Display Type: Built-in Liquid Retina XDR Display
Resolution: 3456 x 2234 Retina
Main Display: Yes
Mirror: Off
Online: Yes
Automatically Adjust Brightness: No
Connection Type: Internal
Installed Packages:
alabaster: 0.7.12
appdirs: 1.4.4
appnope: 0.1.2
Babel: 2.9.1
backcall: 0.2.0
blockdiag: 3.0.0
certifi: 2021.10.8
charset-normalizer: 2.0.12
ChimeraX-AddCharge: 1.2.3
ChimeraX-AddH: 2.1.11
ChimeraX-AlignmentAlgorithms: 2.0
ChimeraX-AlignmentHdrs: 3.2.1
ChimeraX-AlignmentMatrices: 2.0
ChimeraX-Alignments: 2.3
ChimeraX-AlphaFold: 1.0
ChimeraX-AltlocExplorer: 1.0.1
ChimeraX-AmberInfo: 1.0
ChimeraX-Arrays: 1.0
ChimeraX-Atomic: 1.36.3
ChimeraX-AtomicLibrary: 6.1.1
ChimeraX-AtomSearch: 2.0.1
ChimeraX-AxesPlanes: 2.1
ChimeraX-BasicActions: 1.1
ChimeraX-BILD: 1.0
ChimeraX-BlastProtein: 2.0
ChimeraX-BondRot: 2.0
ChimeraX-BugReporter: 1.0
ChimeraX-BuildStructure: 2.6.1
ChimeraX-Bumps: 1.0
ChimeraX-BundleBuilder: 1.1
ChimeraX-ButtonPanel: 1.0
ChimeraX-CageBuilder: 1.0
ChimeraX-CellPack: 1.0
ChimeraX-Centroids: 1.2
ChimeraX-ChemGroup: 2.0
ChimeraX-Clashes: 2.2.2
ChimeraX-ColorActions: 1.0
ChimeraX-ColorGlobe: 1.0
ChimeraX-ColorKey: 1.5.1
ChimeraX-CommandLine: 1.2.2
ChimeraX-ConnectStructure: 2.0.1
ChimeraX-Contacts: 1.0
ChimeraX-Core: 1.4.dev202203271748
ChimeraX-CoreFormats: 1.1
ChimeraX-coulombic: 1.3.2
ChimeraX-Crosslinks: 1.0
ChimeraX-Crystal: 1.0
ChimeraX-CrystalContacts: 1.0
ChimeraX-DataFormats: 1.2.2
ChimeraX-Dicom: 1.1
ChimeraX-DistMonitor: 1.1.5
ChimeraX-Dssp: 2.0
ChimeraX-ExperimentalCommands: 1.0
ChimeraX-FileHistory: 1.0
ChimeraX-FunctionKey: 1.0
ChimeraX-Geometry: 1.1
ChimeraX-gltf: 1.0
ChimeraX-Graphics: 1.1
ChimeraX-Hbonds: 2.1.2
ChimeraX-Help: 1.2
ChimeraX-HKCage: 1.3
ChimeraX-IHM: 1.1
ChimeraX-ImageFormats: 1.2
ChimeraX-IMOD: 1.0
ChimeraX-IO: 1.0.1
ChimeraX-ItemsInspection: 1.0
ChimeraX-Label: 1.1
ChimeraX-ListInfo: 1.1.1
ChimeraX-Log: 1.1.5
ChimeraX-LookingGlass: 1.1
ChimeraX-Maestro: 1.8.1
ChimeraX-Map: 1.1
ChimeraX-MapData: 2.0
ChimeraX-MapEraser: 1.0
ChimeraX-MapFilter: 2.0
ChimeraX-MapFit: 2.0
ChimeraX-MapSeries: 2.1
ChimeraX-Markers: 1.0
ChimeraX-Mask: 1.0
ChimeraX-MatchMaker: 2.0.6
ChimeraX-MDcrds: 2.6
ChimeraX-MedicalToolbar: 1.0.1
ChimeraX-Meeting: 1.0
ChimeraX-MLP: 1.1
ChimeraX-mmCIF: 2.7
ChimeraX-MMTF: 2.1
ChimeraX-Modeller: 1.5.5
ChimeraX-ModelPanel: 1.3.2
ChimeraX-ModelSeries: 1.0
ChimeraX-Mol2: 2.0
ChimeraX-Morph: 1.0
ChimeraX-MouseModes: 1.1
ChimeraX-Movie: 1.0
ChimeraX-Neuron: 1.0
ChimeraX-Nucleotides: 2.0.2
ChimeraX-OpenCommand: 1.8
ChimeraX-PDB: 2.6.6
ChimeraX-PDBBio: 1.0
ChimeraX-PDBLibrary: 1.0.2
ChimeraX-PDBMatrices: 1.0
ChimeraX-PickBlobs: 1.0
ChimeraX-Positions: 1.0
ChimeraX-PresetMgr: 1.1
ChimeraX-PubChem: 2.1
ChimeraX-ReadPbonds: 1.0.1
ChimeraX-Registration: 1.1
ChimeraX-RemoteControl: 1.0
ChimeraX-ResidueFit: 1.0
ChimeraX-RestServer: 1.1
ChimeraX-RNALayout: 1.0
ChimeraX-RotamerLibMgr: 2.0.1
ChimeraX-RotamerLibsDunbrack: 2.0
ChimeraX-RotamerLibsDynameomics: 2.0
ChimeraX-RotamerLibsRichardson: 2.0
ChimeraX-SaveCommand: 1.5
ChimeraX-SchemeMgr: 1.0
ChimeraX-SDF: 2.0
ChimeraX-Segger: 1.0
ChimeraX-Segment: 1.0
ChimeraX-SelInspector: 1.0
ChimeraX-SeqView: 2.5
ChimeraX-Shape: 1.0.1
ChimeraX-Shell: 1.0
ChimeraX-Shortcuts: 1.1
ChimeraX-ShowAttr: 1.0
ChimeraX-ShowSequences: 1.0
ChimeraX-SideView: 1.0
ChimeraX-Smiles: 2.1
ChimeraX-SmoothLines: 1.0
ChimeraX-SpaceNavigator: 1.0
ChimeraX-StdCommands: 1.8
ChimeraX-STL: 1.0
ChimeraX-Storm: 1.0
ChimeraX-StructMeasure: 1.0.1
ChimeraX-Struts: 1.0.1
ChimeraX-Surface: 1.0
ChimeraX-SwapAA: 2.0
ChimeraX-SwapRes: 2.1.1
ChimeraX-TapeMeasure: 1.0
ChimeraX-Test: 1.0
ChimeraX-Toolbar: 1.1
ChimeraX-ToolshedUtils: 1.2.1
ChimeraX-Tug: 1.0
ChimeraX-UI: 1.16.3
ChimeraX-uniprot: 2.2
ChimeraX-UnitCell: 1.0
ChimeraX-ViewDockX: 1.1.2
ChimeraX-VIPERdb: 1.0
ChimeraX-Vive: 1.1
ChimeraX-VolumeMenu: 1.0
ChimeraX-VTK: 1.0
ChimeraX-WavefrontOBJ: 1.0
ChimeraX-WebCam: 1.0
ChimeraX-WebServices: 1.0
ChimeraX-Zone: 1.0
colorama: 0.4.4
cxservices: 1.1
cycler: 0.11.0
Cython: 0.29.26
debugpy: 1.5.1
decorator: 5.1.1
docutils: 0.17.1
entrypoints: 0.4
filelock: 3.4.2
fonttools: 4.31.2
funcparserlib: 1.0.0a0
grako: 3.16.5
html2text: 2020.1.16
idna: 3.3
ihm: 0.27
imagesize: 1.3.0
ipykernel: 6.6.1
ipython: 7.31.1
ipython-genutils: 0.2.0
jedi: 0.18.1
Jinja2: 3.0.3
jupyter-client: 7.1.0
jupyter-core: 4.9.2
kiwisolver: 1.4.0
line-profiler: 3.4.0
lxml: 4.7.1
lz4: 3.1.10
MarkupSafe: 2.1.1
matplotlib: 3.5.1
matplotlib-inline: 0.1.3
msgpack: 1.0.3
nest-asyncio: 1.5.4
networkx: 2.6.3
numpy: 1.22.1
openvr: 1.16.802
packaging: 21.0
ParmEd: 3.4.3
parso: 0.8.3
pexpect: 4.8.0
pickleshare: 0.7.5
Pillow: 9.0.1
pip: 21.3.1
pkginfo: 1.8.2
prompt-toolkit: 3.0.28
psutil: 5.9.0
ptyprocess: 0.7.0
pycollada: 0.7.2
pydicom: 2.2.2
Pygments: 2.11.2
PyOpenGL: 3.1.5
PyOpenGL-accelerate: 3.1.5
pyparsing: 3.0.7
PyQt6: 6.2.3
PyQt6-Qt6: 6.2.4
PyQt6-sip: 13.2.0
PyQt6-WebEngine: 6.2.1
PyQt6-WebEngine-Qt6: 6.2.4
python-dateutil: 2.8.2
pytz: 2022.1
pyzmq: 22.3.0
qtconsole: 5.2.2
QtPy: 2.0.1
requests: 2.27.1
scipy: 1.7.3
setuptools: 59.8.0
six: 1.16.0
snowballstemmer: 2.2.0
sortedcontainers: 2.4.0
Sphinx: 4.3.2
sphinx-autodoc-typehints: 1.15.2
sphinxcontrib-applehelp: 1.0.2
sphinxcontrib-blockdiag: 3.0.0
sphinxcontrib-devhelp: 1.0.2
sphinxcontrib-htmlhelp: 2.0.0
sphinxcontrib-jsmath: 1.0.1
sphinxcontrib-qthelp: 1.0.3
sphinxcontrib-serializinghtml: 1.1.5
suds-community: 1.0.0
tifffile: 2021.11.2
tinyarray: 1.2.4
tornado: 6.1
traitlets: 5.1.1
urllib3: 1.26.9
wcwidth: 0.2.5
webcolors: 1.11.1
wheel: 0.37.1
wheel-filename: 1.3.0
Attachments (1)
{kind=link}
{kind=link}
Change History (4)
comment:1 by , 4 years ago
| Cc: | added |
|---|---|
| Component: | Unassigned → Structure Prediction |
| Owner: | set to |
| Platform: | → all |
| Project: | → ChimeraX |
| Status: | new → assigned |
| Summary: | ChimeraX bug report submission → AlphaFold PAE domains should exclude disordered regions |
| Type: | defect → enhancement |
by , 4 years ago
| Attachment: | bcr_human_pae_domains.png added |
|---|
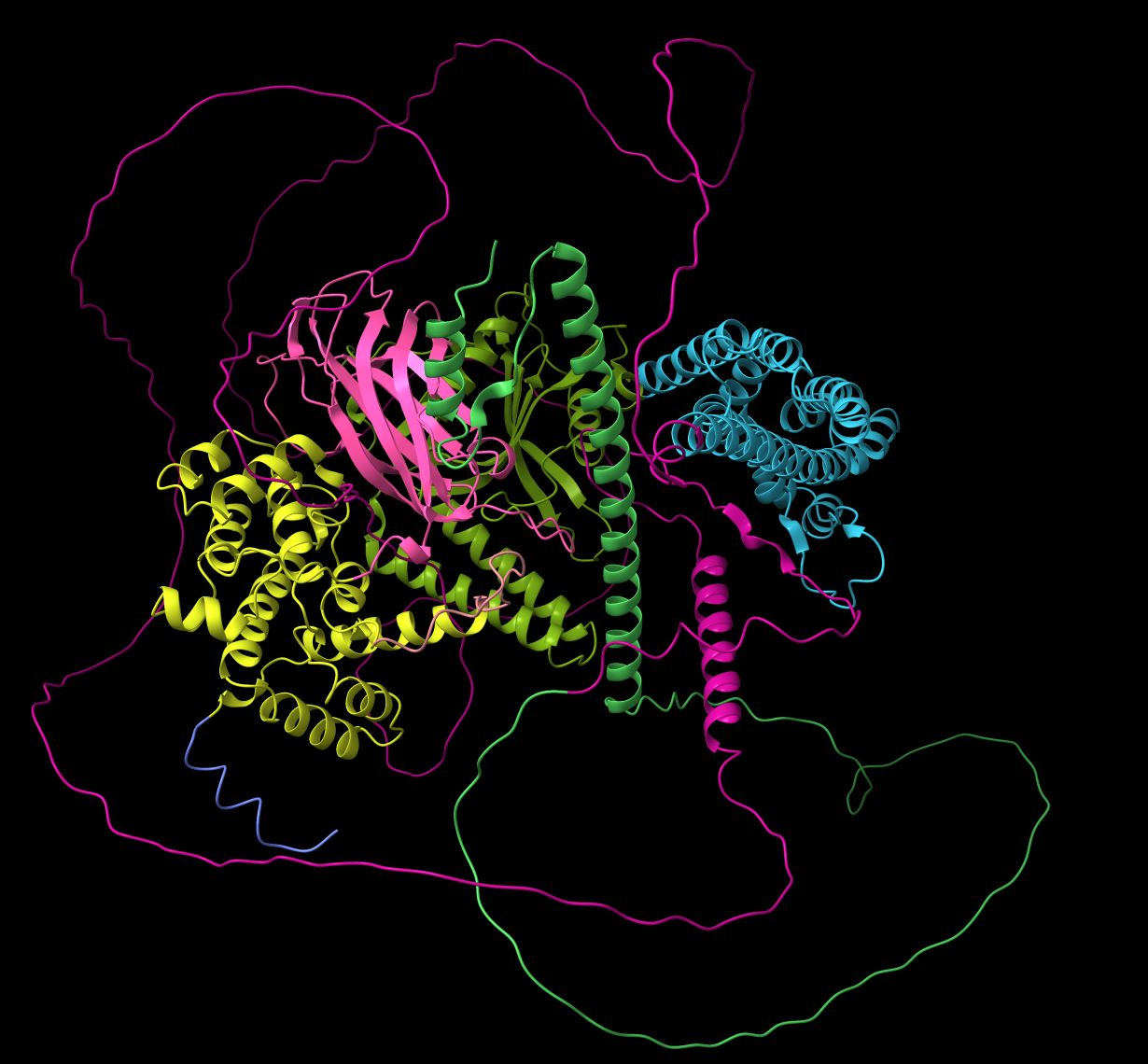
Example of domain coloring included disordered loops.
comment:2 by , 4 years ago
Have to be careful with the interpretation here. There was a lot of discussion about this in the Phenix meeting last week - the upshot is that (to everyone's surprise) isolated secondary structure elements predicted by AlphaFold2 with very low pLDDT are almost always no more real than the unphysical "barbed-wire" (Dave Richardson's term - seems to have caught on) junk coils. Randy Read and Pavel Afonine both did similar experiments - Pavel with a completely random sequence, Randy with the names of all the Phenix contributors interpreted as a protein sequence - and both experiments returned a lot of alpha helices (and even a beta sheet in Randy's case). If you ignore the error estimates and just look at cartoons, the predictions actually look vaguely plausible - but pLDDT values are very low, the PAE matrices show very low confidence even between residues in the same secondary structure element, and showing the solvent-excluded surface reveals exceedingly poor packing.
So the upshot is that in your example, clustering that helix and the two flanking coils into one "domain" is probably reasonable behaviour - but it would certainly be good to clearly flag "domains" like this as junk. Could perhaps rank the domains by mean (median?) pLDDT so the worst ones naturally fall to the bottom? In my current implementation they're just ranked in descending order of size.
comment:3 by , 4 years ago
Interesting. I was wondering about those low confidence isolated alpha-helices. Seems like excluding all residues below a specified pLDDT score (60 or 70) when computing domains might make them more useful. The green domain in the attached image of BCR_HUMAN has 3 helices at the N-terminus that are mostly yellow in pLDDT coloring so score ~70 but are real with solved experimental structure it is an oligomerization domain. But of course there is no way from the scores we are going to be able to perfectly distinguish what is real in the predictions. The aim of this ticket is to make the default domain coloring useful, and give more control if the user wants to fiddle with parameters.
The PAE plots show each residue is pretty strongly connected to the 5 residues before and beyond it in the sequence. I tried another method of excluding edges from the clustering graph that were between nodes <= 5 residues apart. The result was not too bad, but gave some fragmented alpha helices.
It is somewhat surprising that the strong connectivity to previous and following 5 residues does not connect all the residues in the entire sequence. Not really clear how the current algorithm makes the breaks between domains.