Opened 5 years ago
Closed 5 years ago
#5498 closed defect (not a bug)
Discontinuous secondary structure
| Reported by: | Owned by: | Eric Pettersen | |
|---|---|---|---|
| Priority: | normal | Milestone: | |
| Component: | Depiction | Version: | |
| Keywords: | Cc: | ||
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
The following bug report has been submitted:
Platform: macOS-10.16-x86_64-i386-64bit
ChimeraX Version: 1.3.dev202110271145 (2021-10-27 11:45:32 UTC)
Description
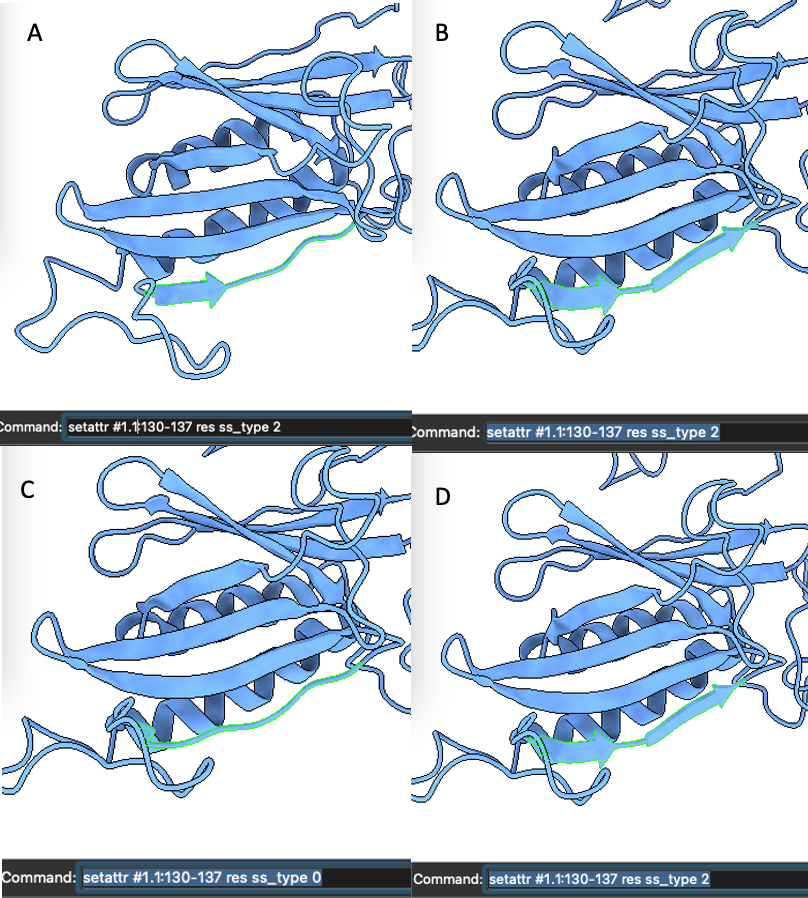
Ribbons do not connect even when forced using e.g., "setattr #1.1:130-137 res ss_type 2". I am trying to beautify a model I have made - it depicts ribbons nicely in PyMOL but ChimeraX shows some as loops. When I use "setattr" encompassing the entire region I want modelled as a continuous beta strand, ChimeraX places several discontinuous ribbons, and i cannot seem to get them connected, even if i set the entire stretch to "type 0"/loop first and then afterwords set the region to "type 2"/sheet (see screen shots - in sequence from A-D).
Log:
UCSF ChimeraX version: 1.3.dev202110271145 (2021-10-27)
© 2016-2021 Regents of the University of California. All rights reserved.
How to cite UCSF ChimeraX
> open "/Volumes/Labs/Kelm/General/Andrea
> Foote/PyMOL:Chimera:Modeling/PurB_2018.pdb" format pdb
Chain information for PurB_2018.pdb #1
---
Chain | Description
A B | No description available
> setattr :243-250 res ss_type 0
Assigning ss_type attribute to 16 items
> setattr :243-250 res ss_type 2
Assigning ss_type attribute to 16 items
> show seq
Expected a collection of one of 'atoms', 'bonds', 'cartoons', 'models',
'pbonds', 'pseudobonds', 'ribbons', or 'surfaces' or a keyword
>
Incomplete command: sequence
> sequence chain
Missing or invalid "chains" argument: empty atom specifier
> A
Unknown command: sequence A
> /A
Unknown command: sequence /A
> sequence chain
Missing or invalid "chains" argument: empty atom specifier
> sequence chain
Missing or invalid "chains" argument: empty atom specifier
> sequence chainn
Missing or invalid "chains" argument: empty atom specifier
> 123angle
123angle is provided by the uninstalled bundle SEQCROW versions 0.13 – 0.15
>
Unknown command: sequence #1
>
Unknown command: sequence #1
> open 5fgp
5fgp title:
Crystal structure of D. melanogaster Pur-alpha repeat I-II in complex with
DNA. [more info...]
Chain information for 5fgp #2
---
Chain | Description | UniProt
A | CG1507-PB, isoform B | Q9V4D9_DROME
B | DNA |
Non-standard residues in 5fgp #2
---
CL — chloride ion
SO4 — sulfate ion
> ui tool show "Show Sequence Viewer"
> sequence chain #1/A
Alignment identifier is 1/A
> ui tool show "Show Sequence Viewer"
> sequence chain #1/A #1/B
Alignment identifier is 1
> ui tool show "Show Sequence Viewer"
> sequence chain #2/A
Alignment identifier is 2/A
> ui tool show "Show Sequence Viewer"
> uniprot
Unknown command: uniprot
> fetch uniprot
Unknown command: fetch uniprot
> select #2
1443 atoms, 1352 bonds, 279 residues, 1 model selected
> uniprot
Unknown command: uniprot
> uniprot 5fgp
Unknown command: uniprot 5fgp
> open uniprot
'uniprot' has no suffix
> open uniprot 5fgp
'uniprot' has no suffix
> open 5fgo
Summary of feedback from opening 5fgo fetched from pdb
---
note | Fetching compressed mmCIF 5fgo from
http://files.rcsb.org/download/5fgo.cif
5fgo title:
Crystal structure of D. melanogaster Pur-alpha repeat III. [more info...]
Chain information for 5fgo #3
---
Chain | Description | UniProt
A B C D E F | CG1507-PB, isoform B | Q9V4D9_DROME
Non-standard residues in 5fgo #3
---
CL — chloride ion
5fgo mmCIF Assemblies
---
1| author_and_software_defined_assembly
2| author_and_software_defined_assembly
3| author_and_software_defined_assembly
> hide sel surfaces
[Repeated 1 time(s)]
> show sel cartoons
> select #3
3471 atoms, 3372 bonds, 553 residues, 1 model selected
> hide sel surfaces
[Repeated 1 time(s)]
> show sel cartoons
> hide sel atoms
> align #2 toAtoms #3
Unequal number of atoms to pair, 1443 and 3471
> show #3 target m
> align #2/AB toAtoms #3
Unequal number of atoms to pair, 0 and 3471
> align #2.1-2 toAtoms #3
Unequal number of atoms to pair, 0 and 3471
> align 2.1-2 and 3
Missing or invalid "atoms" argument: invalid atoms specifier
> align #2 toAtoms #3.1-2
Unequal number of atoms to pair, 1443 and 0
> align #2 toAtoms #3.1
Unequal number of atoms to pair, 1443 and 0
> ~select #3
Nothing selected
> select #3
3471 atoms, 3372 bonds, 553 residues, 1 model selected
> view #3 clip false
> sym #3
5fgo mmCIF Assemblies
---
1| author_and_software_defined_assembly| 1 copy of chains A,B
2| author_and_software_defined_assembly| 1 copy of chains C,D
3| author_and_software_defined_assembly| 1 copy of chains E,F
> log metadata #3
Metadata for 5fgo #3
---
Title | Crystal structure of D. melanogaster Pur-alpha repeat III.
Citation | Weber, J., Bao, H., Hartlmuller, C., Wang, Z., Windhager, A.,
Janowski, R., Madl, T., Jin, P., Niessing, D. (2016). Structural basis of
nucleic-acid recognition and double-strand unwinding by the essential neuronal
protein Pur-alpha. Elife, 5. PMID: 26744780. DOI: 10.7554/eLife.11297
Non-standard residue | CL — chloride ion
Gene source | Drosophila melanogaster (fruit fly)
Experimental method | X-ray diffraction
Resolution | 2.6Å
> log chains #3
Chain information for 5fgo #3
---
Chain | Description | UniProt
A B C D E F | CG1507-PB, isoform B | Q9V4D9_DROME
> align #2/A toAtoms #3.1
Unequal number of atoms to pair, 1274 and 0
> align #2 toAtoms #3
Unequal number of atoms to pair, 1443 and 3471
> align #2 toAtoms #3/AB
Unequal number of atoms to pair, 1443 and 0
> align #2 toAtoms #3/A-B
Unequal number of atoms to pair, 1443 and 1154
> ~select #3
Nothing selected
> hide #2 models
> hide #3 models
> color byattribute chain
No known/registered attribute chain
> split chains
Split PurB_2018.pdb (#1) into 2 models
Split 5fgp (#2) into 2 models
Split 5fgo (#3) into 6 models
Chain information for PurB_2018.pdb A #1.1
---
Chain | Description
A | No description available
Chain information for PurB_2018.pdb B #1.2
---
Chain | Description
B | No description available
Chain information for 5fgp A #2.1
---
Chain | Description
A | No description available
Chain information for 5fgp B #2.2
---
Chain | Description
B | No description available
Chain information for 5fgo A #3.1
---
Chain | Description
A | No description available
Chain information for 5fgo B #3.2
---
Chain | Description
B | No description available
Chain information for 5fgo C #3.3
---
Chain | Description
C | No description available
Chain information for 5fgo D #3.4
---
Chain | Description
D | No description available
Chain information for 5fgo E #3.5
---
Chain | Description
E | No description available
Chain information for 5fgo F #3.6
---
Chain | Description
F | No description available
> hide #1.1 models
> show #1.1 models
> hide #1.2 models
> show #1.2 models
> color #1.1 #dba945
> color #1.1 #db876a
> color #1.2 #7fdba2
> color #1.2 #94b2db
> color #1.2 #639edb
> color #1.1 #e3abdd
> color #1.1 #c666e3
> color #1.1 #e35dd3
> color #1.1 #d5e388
> color #1.1 #d3e36c
> ui mousemode right select
> select clear
> select #1.2/B:42
9 atoms, 8 bonds, 1 residue, 1 model selected
> select add #1.2/B:88
15 atoms, 13 bonds, 2 residues, 1 model selected
> select clear
> select #1.2/B:42
9 atoms, 8 bonds, 1 residue, 1 model selected
> select #1.2/B:89
8 atoms, 7 bonds, 1 residue, 1 model selected
> select #1.2/B:43
7 atoms, 6 bonds, 1 residue, 1 model selected
> select add #1.2/B:88
13 atoms, 11 bonds, 2 residues, 1 model selected
> select clear
[Repeated 6 time(s)]
> select #1.2/B:44
9 atoms, 8 bonds, 1 residue, 1 model selected
> select #1.2/B:88
6 atoms, 5 bonds, 1 residue, 1 model selected
> select clear
[Repeated 1 time(s)]
> select #1.2/B:65
8 atoms, 7 bonds, 1 residue, 1 model selected
> select up
72 atoms, 73 bonds, 8 residues, 1 model selected
> select up
2390 atoms, 2433 bonds, 324 residues, 1 model selected
> select down
72 atoms, 73 bonds, 8 residues, 1 model selected
> select down
8 atoms, 7 bonds, 1 residue, 1 model selected
> select up
72 atoms, 73 bonds, 8 residues, 1 model selected
> select add #1.2/B:68
76 atoms, 76 bonds, 9 residues, 1 model selected
> select up
148 atoms, 149 bonds, 18 residues, 1 model selected
> select #1.2/B:78
5 atoms, 4 bonds, 1 residue, 1 model selected
> select clear
> select #1.2/B:46
8 atoms, 7 bonds, 1 residue, 1 model selected
> select up
277 atoms, 281 bonds, 46 residues, 1 model selected
> select down
8 atoms, 7 bonds, 1 residue, 1 model selected
> select down
8 atoms, 7 bonds, 1 residue, 1 model selected
> select down
8 atoms, 7 bonds, 1 residue, 1 model selected
> select up
277 atoms, 281 bonds, 46 residues, 1 model selected
> select down
8 atoms, 7 bonds, 1 residue, 1 model selected
> select add #1.2/B:47
13 atoms, 11 bonds, 2 residues, 1 model selected
> select up
316 atoms, 320 bonds, 51 residues, 1 model selected
> select down
13 atoms, 11 bonds, 2 residues, 1 model selected
> select up
316 atoms, 320 bonds, 51 residues, 1 model selected
> select down
13 atoms, 11 bonds, 2 residues, 1 model selected
> select add #1.2/B:51
21 atoms, 18 bonds, 3 residues, 1 model selected
> select up
316 atoms, 320 bonds, 51 residues, 1 model selected
> select down
21 atoms, 18 bonds, 3 residues, 1 model selected
> select clear
> select #1.2/B:47
5 atoms, 4 bonds, 1 residue, 1 model selected
> select up
39 atoms, 38 bonds, 5 residues, 1 model selected
> select #1.2/B:52
8 atoms, 7 bonds, 1 residue, 1 model selected
> select #1.2/B:47
5 atoms, 4 bonds, 1 residue, 1 model selected
> select up
39 atoms, 38 bonds, 5 residues, 1 model selected
> select add #1.2/B:52
47 atoms, 45 bonds, 6 residues, 1 model selected
> select up
92 atoms, 91 bonds, 11 residues, 1 model selected
> select add #1.2/B:58
103 atoms, 102 bonds, 12 residues, 1 model selected
> select up
164 atoms, 165 bonds, 19 residues, 1 model selected
> select up
2390 atoms, 2433 bonds, 324 residues, 1 model selected
> select down
164 atoms, 165 bonds, 19 residues, 1 model selected
> select subtract #1.2/B:65
156 atoms, 158 bonds, 18 residues, 1 model selected
> select add #1.2/B:65
164 atoms, 165 bonds, 19 residues, 1 model selected
> select add #1.2/B:66
169 atoms, 169 bonds, 20 residues, 1 model selected
> select up
178 atoms, 179 bonds, 21 residues, 1 model selected
> select add #1.2/B:68
182 atoms, 182 bonds, 22 residues, 1 model selected
> select up
254 atoms, 256 bonds, 31 residues, 1 model selected
> select add #1.2/B:78
259 atoms, 260 bonds, 32 residues, 1 model selected
> select up
267 atoms, 269 bonds, 34 residues, 1 model selected
> select add #1.2/B:81
273 atoms, 274 bonds, 35 residues, 1 model selected
> select up
328 atoms, 330 bonds, 42 residues, 1 model selected
> select add #1.2/B:89
336 atoms, 337 bonds, 43 residues, 1 model selected
> select up
478 atoms, 484 bonds, 61 residues, 1 model selected
> select add #1.2/B:46
486 atoms, 491 bonds, 62 residues, 1 model selected
> select add #1.2/B:45
495 atoms, 499 bonds, 63 residues, 1 model selected
> select add #1.2/B:44
504 atoms, 507 bonds, 64 residues, 1 model selected
> select add #1.2/B:43
511 atoms, 513 bonds, 65 residues, 1 model selected
> select add #1.2/B:42
520 atoms, 521 bonds, 66 residues, 1 model selected
> select add #1.2/B:108
529 atoms, 529 bonds, 67 residues, 1 model selected
> select add #1.2/B:109
537 atoms, 536 bonds, 68 residues, 1 model selected
> create II sele
Unknown command: create II sele
Drag select of 84 residues
Drag select of 24 residues
> select clear
Drag select of 11 residues
Drag select of 12 residues
> select up
531 atoms, 538 bonds, 77 residues, 1 model selected
Drag select of 37 residues
> select subtract #1.1/A:19
321 atoms, 535 bonds, 39 residues, 1 model selected
> select subtract #1.1/A:20
314 atoms, 528 bonds, 38 residues, 1 model selected
> select subtract #1.1/A:40
305 atoms, 520 bonds, 37 residues, 1 model selected
> select subtract #1.1/A:41
296 atoms, 512 bonds, 36 residues, 1 model selected
> select add #1.1/A:78
301 atoms, 516 bonds, 37 residues, 1 model selected
> select up
515 atoms, 517 bonds, 78 residues, 1 model selected
> select down
301 atoms, 516 bonds, 37 residues, 1 model selected
> select add #1.1/A:79
305 atoms, 519 bonds, 38 residues, 1 model selected
> select add #1.1/A:80
309 atoms, 522 bonds, 39 residues, 1 model selected
> select add #1.1/A:81
315 atoms, 527 bonds, 40 residues, 1 model selected
> select up
529 atoms, 531 bonds, 81 residues, 1 model selected
> select down
315 atoms, 527 bonds, 40 residues, 1 model selected
> select add #1.1/A:82
324 atoms, 535 bonds, 41 residues, 1 model selected
> select up
538 atoms, 540 bonds, 82 residues, 1 model selected
> select down
324 atoms, 535 bonds, 41 residues, 1 model selected
Drag select of 4 residues
> select add #1.1/A:82
349 atoms, 535 bonds, 44 residues, 1 model selected
> select add #1.1/A:86
356 atoms, 541 bonds, 45 residues, 1 model selected
> select add #1.1/A:87
364 atoms, 548 bonds, 46 residues, 1 model selected
> select add #1.1/A:88
370 atoms, 553 bonds, 47 residues, 1 model selected
> show target m
[Repeated 1 time(s)]
> hide target m
> show #!1 models
> show #1.1 models
> hide #!1 models
> show #!1 models
Drag select of 5 residues
> select add #1.1/A:88
393 atoms, 553 bonds, 51 residues, 1 model selected
> select add #1.1/A:108
402 atoms, 561 bonds, 52 residues, 1 model selected
> select clear
> ui tool show "Blast Protein"
> ui tool show "Model Loops"
> ui tool show "Modeller Comparative"
> ui tool show "Show Sequence Viewer"
> sequence chain #2.1/A
Alignment identifier is 2.1/A
> select #2.1/A:41
9 atoms, 8 bonds, 1 residue, 1 model selected
> select #2.1/A:41-52
98 atoms, 97 bonds, 12 residues, 1 model selected
> ui tool show "Show Sequence Viewer"
> sequence chain #1.1/A
Alignment identifier is 1.1/A
> select #1.1/A:2
5 atoms, 4 bonds, 1 residue, 1 model selected
> select #1.1/A:2-5
25 atoms, 24 bonds, 4 residues, 1 model selected
> select #1.1/A:2-5,9-17
73 atoms, 71 bonds, 13 residues, 1 model selected
> select #1.1/A:2-5,9-17,23-33
143 atoms, 143 bonds, 24 residues, 1 model selected
> select clear
> select #1.1/A:19-20
11 atoms, 11 bonds, 2 residues, 1 model selected
> select #1.1/A:19-24
39 atoms, 40 bonds, 6 residues, 1 model selected
> select #1.1/A:28
11 atoms, 10 bonds, 1 residue, 1 model selected
> select #1.1/A:28-36
43 atoms, 42 bonds, 9 residues, 1 model selected
> select clear
[Repeated 2 time(s)]
> select #1.1/A:155
11 atoms, 11 bonds, 1 residue, 1 model selected
> select #1.1/A:155-164
91 atoms, 91 bonds, 10 residues, 1 model selected
> select #1.1/A:19-24
39 atoms, 40 bonds, 6 residues, 1 model selected
> select #1.1/A:27-28
18 atoms, 18 bonds, 2 residues, 1 model selected
> select #1.1/A:27-32
34 atoms, 34 bonds, 6 residues, 1 model selected
> select #1.1/A:38
4 atoms, 3 bonds, 1 residue, 1 model selected
> select #1.1/A:38-44
51 atoms, 50 bonds, 7 residues, 1 model selected
> select #1.1/A:27-32
34 atoms, 34 bonds, 6 residues, 1 model selected
> select #1.1/A:19-24
39 atoms, 40 bonds, 6 residues, 1 model selected
> ui tool show "Show Sequence Viewer"
> sequence chain #1.1/A
Destroying pre-existing alignment with identifier 1.1/A
Alignment identifier is 1.1/A
> select #1.1/A:10
11 atoms, 10 bonds, 1 residue, 1 model selected
> select #1.1/A:10-16
35 atoms, 34 bonds, 7 residues, 1 model selected
> select #1.1/A:22-23
15 atoms, 15 bonds, 2 residues, 1 model selected
> select #1.1/A:22-32
70 atoms, 72 bonds, 11 residues, 1 model selected
> select #1.1/A:10-16
35 atoms, 34 bonds, 7 residues, 1 model selected
> select #1.1/A:22-32
70 atoms, 72 bonds, 11 residues, 1 model selected
> select clear
[Repeated 1 time(s)]
> select #1.1/A:117-118
13 atoms, 12 bonds, 2 residues, 1 model selected
> select #1.1/A:117-127
64 atoms, 64 bonds, 11 residues, 1 model selected
> select #1.1/A:10
11 atoms, 10 bonds, 1 residue, 1 model selected
> select #1.1/A:10-11
15 atoms, 14 bonds, 2 residues, 1 model selected
> select clear
> select #1.1/A:166
4 atoms, 3 bonds, 1 residue, 1 model selected
> select #1.1/A:166-168
12 atoms, 11 bonds, 3 residues, 1 model selected
> select #1.1/A:10-16
35 atoms, 34 bonds, 7 residues, 1 model selected
> set bgColor white
> color #1.1 #75bee6
> color #1.1 #70a9ff
> show sel surfaces
> select clear
> show #!1.1 surfaces
> transparency #1.1#!1 100
> graphics silhouettes true
> lighting soft
> graphics silhouettes false
> graphics silhouettes true
> lighting simple
[Repeated 1 time(s)]
> lighting soft
> lighting full
> lighting flat
> lighting shadows true intensity 0.5
> graphics silhouettes false
> select #1.1/A:15@O
1 atom, 1 residue, 1 model selected
> select clear
> hide #1.1.1 models
> show #1.1.1 models
> graphics silhouettes true
> hide #!1.1 models
> show #!1.1 models
> select #1.1.1
2390 atoms, 324 residues, 1 model selected
> ~select #1.1.1
1 model selected
> select #1.1
2390 atoms, 2433 bonds, 324 residues, 1 model selected
> ~select #1.1.1
2433 bonds, 2 models selected
> graphics silhouettes false
> select #1.1.1
2390 atoms, 324 residues, 1 model selected
> select #1.1
2390 atoms, 2433 bonds, 324 residues, 1 model selected
> ~select #1.1
1 model selected
> select #1.1.1
2390 atoms, 324 residues, 1 model selected
> graphics silhouettes true
> select clear
> lighting flat
> lighting full
> lighting soft
> select #1.1.1
2390 atoms, 324 residues, 1 model selected
> select #1.1
2390 atoms, 2433 bonds, 324 residues, 1 model selected
> graphics silhouettes false
> ~select #1.1
1 model selected
> graphics silhouettes true
> graphics silhouettes false
> select #1.1.1
2390 atoms, 324 residues, 1 model selected
> graphics silhouettes true
> ~select #1.1.1
1 model selected
> select #1.1.1
2390 atoms, 324 residues, 1 model selected
> show #1.1.1 target m
> hide #1.1.1 target m
[Repeated 1 time(s)]
> show #1.1.1 target m
> hide #!1.1 models
> show #!1.1 models
> select #1.1
2390 atoms, 2433 bonds, 324 residues, 1 model selected
> ~select #1.1.1
2433 bonds, 2 models selected
> select #1.1
2390 atoms, 2433 bonds, 324 residues, 1 model selected
> hide #1.1.1 target m
> show #1.1.1 models
> hide #!1.1 target m
> show #!1.1 target m
> tube helices
Unknown command: tube helices
> cylinders
Unknown command: cylinders
> ~surface
> surface
> ~cartoon
> cartoon
> select 1.1:1-210
Expected an objects specifier or a keyword
> select #1.1:1-210
1530 atoms, 1554 bonds, 210 residues, 1 model selected
> sticks
Unknown command: sticks
> spheres
Unknown command: spheres
> ~surface
> surface and sele
Expected an atoms specifier or a keyword
> surface sele
Expected an atoms specifier or a keyword
> ~select
Nothing selected
> ~select
Nothing selected
> select
9694 atoms, 9590 bonds, 1480 residues, 13 models selected
> select up
9694 atoms, 9590 bonds, 1480 residues, 23 models selected
> select down
13 models selected
> select up
3 models selected
> select up
3 models selected
> select up
3 models selected
> select up
3 models selected
> select up
3 models selected
> select down
3 models selected
> select down
3 models selected
> select down
3 models selected
> select down
3 models selected
> select down
3 models selected
> select down
3 models selected
> select down
3 models selected
> select up
3 models selected
> select up
3 models selected
> select up
3 models selected
> select up
3 models selected
> select up
3 models selected
> select up
3 models selected
> select up
3 models selected
> select up
3 models selected
> select up
3 models selected
> select up
3 models selected
> select up
3 models selected
> select #2
1443 atoms, 1352 bonds, 279 residues, 3 models selected
> select #1
4780 atoms, 4866 bonds, 648 residues, 3 models selected
> ~select #1.2
2390 atoms, 2433 bonds, 324 residues, 4 models selected
> select clear
> select #1.1/A:197
5 atoms, 4 bonds, 1 residue, 1 model selected
> select #1.1/A:201
8 atoms, 7 bonds, 1 residue, 1 model selected
> select #1.1/A:200
9 atoms, 8 bonds, 1 residue, 1 model selected
> select clear
Drag select of 219 residues
> select clear
> help
> show #!2.1 models
> show #!2.2 models
> show #!3 models
> hide #!2 models
> hide #!2.1 models
> hide #!2.2 models
> select #1.1/A:230
9 atoms, 8 bonds, 1 residue, 1 model selected
> select add #1.1/A:232
16 atoms, 15 bonds, 2 residues, 2 models selected
> select add #1.1/A:231
24 atoms, 22 bonds, 3 residues, 2 models selected
> select add #1.1/A:233
33 atoms, 30 bonds, 4 residues, 2 models selected
> select add #1.1/A:234
37 atoms, 33 bonds, 5 residues, 2 models selected
> select add #1.1/A:235
44 atoms, 39 bonds, 6 residues, 2 models selected
> select add #1.1/A:236
50 atoms, 44 bonds, 7 residues, 2 models selected
> select up
230 atoms, 234 bonds, 36 residues, 2 models selected
> select up
2390 atoms, 2433 bonds, 324 residues, 2 models selected
> select up
2390 atoms, 2433 bonds, 324 residues, 2 models selected
> select up
4780 atoms, 4866 bonds, 648 residues, 3 models selected
> select down
2390 atoms, 2433 bonds, 324 residues, 3 models selected
> select down
1 model selected
> select down
230 atoms, 234 bonds, 36 residues, 1 model selected
> select down
50 atoms, 44 bonds, 7 residues, 2 models selected
> select down
50 atoms, 44 bonds, 7 residues, 2 models selected
> select down
50 atoms, 44 bonds, 7 residues, 2 models selected
> hide #!3 models
> show #!3 models
> hide #!1 models
> show #!3.1 models
> show #!3.2 models
> show #!3.3 models
> show #!3.4 models
> show #!3.5 models
> show #!3.6 models
> hide #!3.3 models
> hide #!3.4 models
> hide #!3.5 models
> hide #!3.6 models
> hide #!3.1 models
> hide #!3 models
> show #!3 models
> hide #!1.1 models
> hide #1.1.1 models
> select #3.2/B:200
9 atoms, 8 bonds, 1 residue, 1 model selected
> select #3.2/B:192
8 atoms, 7 bonds, 1 residue, 1 model selected
> setattr #3.2:192-200 res ss_type 2
Assigning ss_type attribute to 9 items
> hide #!3 models
> hide #!3.2 models
> show #1.1.1 models
> hide #1.1.1 models
> select #1.1/A:133
6 atoms, 5 bonds, 1 residue, 1 model selected
> select clear
> select #1.1/A:130
5 atoms, 4 bonds, 1 residue, 1 model selected
> select add #1.1/A:131
13 atoms, 11 bonds, 2 residues, 2 models selected
> select add #1.1/A:132
22 atoms, 19 bonds, 3 residues, 2 models selected
> select add #1.1/A:133
28 atoms, 24 bonds, 4 residues, 2 models selected
> select add #1.1/A:134
37 atoms, 32 bonds, 5 residues, 2 models selected
> select add #1.1/A:135
48 atoms, 43 bonds, 6 residues, 2 models selected
> select add #1.1/A:136
56 atoms, 50 bonds, 7 residues, 2 models selected
> select add #1.1/A:137
63 atoms, 56 bonds, 8 residues, 2 models selected
> setattr #1.1:130-137 res ss_type 2
Assigning ss_type attribute to 8 items
> setattr #1.1:130-137 res ss_type 0
Assigning ss_type attribute to 8 items
> setattr #1.1:130-137 res ss_type 2
Assigning ss_type attribute to 8 items
OpenGL version: 4.1 INTEL-16.1.12
OpenGL renderer: Intel(R) Iris(TM) Graphics 6100
OpenGL vendor: Intel Inc.Hardware:
Hardware Overview:
Model Name: MacBook Pro
Model Identifier: MacBookPro12,1
Processor Name: Dual-Core Intel Core i5
Processor Speed: 2.7 GHz
Number of Processors: 1
Total Number of Cores: 2
L2 Cache (per Core): 256 KB
L3 Cache: 3 MB
Hyper-Threading Technology: Enabled
Memory: 16 GB
System Firmware Version: 426.0.0.0.0
SMC Version (system): 2.28f7
Software:
System Software Overview:
System Version: macOS 11.2.3 (20D91)
Kernel Version: Darwin 20.3.0
Time since boot: 1 day 8 minutes
Graphics/Displays:
Intel Iris Graphics 6100:
Chipset Model: Intel Iris Graphics 6100
Type: GPU
Bus: Built-In
VRAM (Dynamic, Max): 1536 MB
Vendor: Intel
Device ID: 0x162b
Revision ID: 0x0009
Metal Family: Supported, Metal GPUFamily macOS 1
Displays:
LED Cinema Display:
Display Type: LCD
Resolution: 2048 x 1152
UI Looks like: 2048 x 1152
Framebuffer Depth: 30-Bit Color (ARGB2101010)
Display Serial Number: 2A04244C6JL
Main Display: Yes
Mirror: Off
Online: Yes
Rotation: Supported
Automatically Adjust Brightness: Yes
Connection Type: Thunderbolt/DisplayPort
Locale: (None, 'UTF-8')
PyQt5 5.15.2, Qt 5.15.2
Installed Packages:
alabaster: 0.7.12
appdirs: 1.4.4
appnope: 0.1.2
Babel: 2.9.1
backcall: 0.2.0
blockdiag: 2.0.1
certifi: 2021.5.30
cftime: 1.5.1
charset-normalizer: 2.0.7
ChimeraX-AddCharge: 1.1.4
ChimeraX-AddH: 2.1.10
ChimeraX-AlignmentAlgorithms: 2.0
ChimeraX-AlignmentHdrs: 3.2
ChimeraX-AlignmentMatrices: 2.0
ChimeraX-Alignments: 2.2.3
ChimeraX-AlphaFold: 1.0
ChimeraX-AltlocExplorer: 1.0.1
ChimeraX-AmberInfo: 1.0
ChimeraX-Arrays: 1.0
ChimeraX-Atomic: 1.31
ChimeraX-AtomicLibrary: 4.1.6
ChimeraX-AtomSearch: 2.0
ChimeraX-AtomSearchLibrary: 1.0
ChimeraX-AxesPlanes: 2.1
ChimeraX-BasicActions: 1.1
ChimeraX-BILD: 1.0
ChimeraX-BlastProtein: 2.0
ChimeraX-BondRot: 2.0
ChimeraX-BugReporter: 1.0
ChimeraX-BuildStructure: 2.6
ChimeraX-Bumps: 1.0
ChimeraX-BundleBuilder: 1.1
ChimeraX-ButtonPanel: 1.0
ChimeraX-CageBuilder: 1.0
ChimeraX-CellPack: 1.0
ChimeraX-Centroids: 1.2
ChimeraX-ChemGroup: 2.0
ChimeraX-Clashes: 2.2.1
ChimeraX-ColorActions: 1.0
ChimeraX-ColorGlobe: 1.0
ChimeraX-ColorKey: 1.5
ChimeraX-CommandLine: 1.1.5
ChimeraX-ConnectStructure: 2.0
ChimeraX-Contacts: 1.0
ChimeraX-Core: 1.3.dev202110271145
ChimeraX-CoreFormats: 1.1
ChimeraX-coulombic: 1.3.1
ChimeraX-Crosslinks: 1.0
ChimeraX-Crystal: 1.0
ChimeraX-CrystalContacts: 1.0
ChimeraX-DataFormats: 1.2.2
ChimeraX-Dicom: 1.0
ChimeraX-DistMonitor: 1.1.5
ChimeraX-Dssp: 2.0
ChimeraX-EMDB-SFF: 1.0
ChimeraX-ExperimentalCommands: 1.0
ChimeraX-FileHistory: 1.0
ChimeraX-FunctionKey: 1.0
ChimeraX-Geometry: 1.1
ChimeraX-gltf: 1.0
ChimeraX-Graphics: 1.1
ChimeraX-Hbonds: 2.1.2
ChimeraX-Help: 1.2
ChimeraX-HKCage: 1.3
ChimeraX-IHM: 1.1
ChimeraX-ImageFormats: 1.2
ChimeraX-IMOD: 1.0
ChimeraX-IO: 1.0.1
ChimeraX-ItemsInspection: 1.0
ChimeraX-Label: 1.1
ChimeraX-ListInfo: 1.1.1
ChimeraX-Log: 1.1.4
ChimeraX-LookingGlass: 1.1
ChimeraX-Maestro: 1.8.1
ChimeraX-Map: 1.1
ChimeraX-MapData: 2.0
ChimeraX-MapEraser: 1.0
ChimeraX-MapFilter: 2.0
ChimeraX-MapFit: 2.0
ChimeraX-MapSeries: 2.1
ChimeraX-Markers: 1.0
ChimeraX-Mask: 1.0
ChimeraX-MatchMaker: 2.0.3
ChimeraX-MDcrds: 2.6
ChimeraX-MedicalToolbar: 1.0.1
ChimeraX-Meeting: 1.0
ChimeraX-MLP: 1.1
ChimeraX-mmCIF: 2.4
ChimeraX-MMTF: 2.1
ChimeraX-Modeller: 1.2.4
ChimeraX-ModelPanel: 1.2
ChimeraX-ModelSeries: 1.0
ChimeraX-Mol2: 2.0
ChimeraX-Morph: 1.0
ChimeraX-MouseModes: 1.1
ChimeraX-Movie: 1.0
ChimeraX-Neuron: 1.0
ChimeraX-Nucleotides: 2.0.2
ChimeraX-OpenCommand: 1.8
ChimeraX-PDB: 2.6.4
ChimeraX-PDBBio: 1.0
ChimeraX-PDBLibrary: 1.0.2
ChimeraX-PDBMatrices: 1.0
ChimeraX-PickBlobs: 1.0
ChimeraX-Positions: 1.0
ChimeraX-PresetMgr: 1.0.1
ChimeraX-PubChem: 2.1
ChimeraX-ReadPbonds: 1.0
ChimeraX-Registration: 1.1
ChimeraX-RemoteControl: 1.0
ChimeraX-ResidueFit: 1.0
ChimeraX-RestServer: 1.1
ChimeraX-RNALayout: 1.0
ChimeraX-RotamerLibMgr: 2.0
ChimeraX-RotamerLibsDunbrack: 2.0
ChimeraX-RotamerLibsDynameomics: 2.0
ChimeraX-RotamerLibsRichardson: 2.0
ChimeraX-SaveCommand: 1.5
ChimeraX-SchemeMgr: 1.0
ChimeraX-SDF: 2.0
ChimeraX-Segger: 1.0
ChimeraX-Segment: 1.0
ChimeraX-SelInspector: 1.0
ChimeraX-SeqView: 2.4.5
ChimeraX-Shape: 1.0.1
ChimeraX-Shell: 1.0
ChimeraX-Shortcuts: 1.1
ChimeraX-ShowAttr: 1.0
ChimeraX-ShowSequences: 1.0
ChimeraX-SideView: 1.0
ChimeraX-Smiles: 2.1
ChimeraX-SmoothLines: 1.0
ChimeraX-SpaceNavigator: 1.0
ChimeraX-StdCommands: 1.6
ChimeraX-STL: 1.0
ChimeraX-Storm: 1.0
ChimeraX-StructMeasure: 1.0
ChimeraX-Struts: 1.0
ChimeraX-Surface: 1.0
ChimeraX-SwapAA: 2.0
ChimeraX-SwapRes: 2.1
ChimeraX-TapeMeasure: 1.0
ChimeraX-Test: 1.0
ChimeraX-Toolbar: 1.1
ChimeraX-ToolshedUtils: 1.2
ChimeraX-Tug: 1.0
ChimeraX-UI: 1.13.6
ChimeraX-uniprot: 2.2
ChimeraX-UnitCell: 1.0
ChimeraX-ViewDockX: 1.0.1
ChimeraX-VIPERdb: 1.0
ChimeraX-Vive: 1.1
ChimeraX-VolumeMenu: 1.0
ChimeraX-VTK: 1.0
ChimeraX-WavefrontOBJ: 1.0
ChimeraX-WebCam: 1.0
ChimeraX-WebServices: 1.0
ChimeraX-Zone: 1.0
colorama: 0.4.4
cxservices: 1.1
cycler: 0.10.0
Cython: 0.29.24
decorator: 5.1.0
docutils: 0.17.1
filelock: 3.0.12
funcparserlib: 0.3.6
grako: 3.16.5
h5py: 3.5.0
html2text: 2020.1.16
idna: 3.3
ihm: 0.21
imagecodecs: 2021.4.28
imagesize: 1.2.0
ipykernel: 5.5.5
ipython: 7.23.1
ipython-genutils: 0.2.0
jedi: 0.18.0
Jinja2: 3.0.1
jupyter-client: 6.1.12
jupyter-core: 4.9.0
kiwisolver: 1.3.2
lxml: 4.6.3
lz4: 3.1.3
MarkupSafe: 2.0.1
matplotlib: 3.4.3
matplotlib-inline: 0.1.3
msgpack: 1.0.2
netCDF4: 1.5.7
networkx: 2.6.3
numexpr: 2.7.3
numpy: 1.21.2
openvr: 1.16.801
packaging: 21.0
ParmEd: 3.2.0
parso: 0.8.2
pexpect: 4.8.0
pickleshare: 0.7.5
Pillow: 8.3.2
pip: 21.2.4
pkginfo: 1.7.1
prompt-toolkit: 3.0.21
psutil: 5.8.0
ptyprocess: 0.7.0
pycollada: 0.7.1
pydicom: 2.1.2
Pygments: 2.10.0
PyOpenGL: 3.1.5
PyOpenGL-accelerate: 3.1.5
pyparsing: 3.0.2
PyQt5-commercial: 5.15.2
PyQt5-sip: 12.8.1
PyQtWebEngine-commercial: 5.15.2
python-dateutil: 2.8.2
pytz: 2021.3
pyzmq: 22.3.0
qtconsole: 5.1.1
QtPy: 1.11.2
RandomWords: 0.3.0
requests: 2.26.0
scipy: 1.7.1
setuptools: 57.5.0
sfftk-rw: 0.7.1
six: 1.16.0
snowballstemmer: 2.1.0
sortedcontainers: 2.4.0
Sphinx: 4.2.0
sphinx-autodoc-typehints: 1.12.0
sphinxcontrib-applehelp: 1.0.2
sphinxcontrib-blockdiag: 2.0.0
sphinxcontrib-devhelp: 1.0.2
sphinxcontrib-htmlhelp: 2.0.0
sphinxcontrib-jsmath: 1.0.1
sphinxcontrib-qthelp: 1.0.3
sphinxcontrib-serializinghtml: 1.1.5
suds-jurko: 0.6
tifffile: 2021.4.8
tinyarray: 1.2.3
tornado: 6.1
traitlets: 5.1.1
urllib3: 1.26.7
wcwidth: 0.2.5
webcolors: 1.11.1
wheel: 0.37.0
wheel-filename: 1.3.0
File attachment: Screen Shot 2021-10-28 at 1.36.09 PM.png
{kind=link}
{kind=link}
Attachments (1)
Change History (3)
by , 5 years ago
| Attachment: | Screen Shot 2021-10-28 at 1.36.09 PM.png added |
|---|
comment:1 by , 5 years ago
| Component: | Unassigned → Depiction |
|---|---|
| Owner: | set to |
| Platform: | → all |
| Project: | → ChimeraX |
| Status: | new → accepted |
| Summary: | ChimeraX bug report submission → Discontinuous secondary structure |
comment:2 by , 5 years ago
| Resolution: | → not a bug |
|---|---|
| Status: | accepted → closed |
Hi Andrea,
In order to handle the case where a structure has adjacent helices with no intervening non-helix residue, there is also an 'ss_id' attribute to mark what secondary structure element a residue belongs to, and there will always be a short coil depiction between residues with differing ss_ids. So to get the effect you want to achieve you also have to assign the same ss_id to all those residues. I would suggest assigning an ss_id that isn't likely to already be used, such as 101, 102, etc.
--Eric
Eric Pettersen
UCSF Computer Graphics Lab
Note:
See TracTickets
for help on using tickets.
Added by email2trac