#4861 closed defect (fixed)
Failure running antechamber on Windows
| Reported by: | Owned by: | Eric Pettersen | |
|---|---|---|---|
| Priority: | normal | Milestone: | |
| Component: | Structure Editing | Version: | |
| Keywords: | Cc: | chimera-programmers | |
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
The following bug report has been submitted:
Platform: Windows-10-10.0.19041
ChimeraX Version: 1.2.5 (2021-05-24 04:13:57 UTC)
Description
coulombicUsing Amber 20 recommended default charges and atom types for standard residues
Assigning partial charges to residue HIC (net charge +0) with am1-bcc method
Running ANTECHAMBER command: C:/Program Files/ChimeraX 1.2.5/bin/amber20/bin/antechamber -ek qm_theory='AM1', -i C:\Users\myHP\AppData\Local\Temp\tmphr9qv0v0\ante.in.mol2 -fi mol2 -o C:\Users\myHP\AppData\Local\Temp\tmphr9qv0v0\ante.out.mol2 -fo mol2 -c bcc -nc 0 -j 5 -s 2 -dr n
(HIC)
(HIC) Welcome to antechamber 20.0: molecular input file processor.
(HIC)
(HIC) Info: Finished reading file (C:\Users\myHP\AppData\Local\Temp\tmphr9qv0v0\ante.in.mol2); atoms read (32), bonds read (32).
(HIC) Info: Determining atomic numbers from atomic symbols which are case sensitive.
(HIC) Running: C:/Program Files/ChimeraX 1.2.5/bin/amber20/bin/bondtype -j part -i ANTECHAMBER_BOND_TYPE.AC0 -o ANTECHAMBER_BOND_TYPE.AC -f ac
(HIC) /usr/bin/antechamber: Fatal Error!
(HIC) Cannot properly run "C:/Program Files/ChimeraX 1.2.5/bin/amber20/bin/bondtype -j part -i ANTECHAMBER_BOND_TYPE.AC0 -o ANTECHAMBER_BOND_TYPE.AC -f ac".
(HIC) No such file or directory
Failure running ANTECHAMBER for residue HIC Check reply log for details
Log:
UCSF ChimeraX version: 1.2.5 (2021-05-24)
© 2016-2021 Regents of the University of California. All rights reserved.
How to cite UCSF ChimeraX
> open "D:\user\Box Sync\manuscripts\Dorit lab yeat WT and 167 mutant
> cryoEM\actin structure files Niels Volkmann shared on 21-01-19\wt_model.pdb"
> format pdb
Summary of feedback from opening D:\user\Box Sync\manuscripts\Dorit lab yeat
WT and 167 mutant cryoEM\actin structure files Niels Volkmann shared on
21-01-19\wt_model.pdb
---
warnings | Ignored bad PDB record found on line 25
GEOMETRY RESTRAINTS LIBRARY: CDL v1.2
Ignored bad PDB record found on line 26
DEVIATIONS FROM IDEAL VALUES.
Ignored bad PDB record found on line 27
BOND : 0.006 0.054 14780
Ignored bad PDB record found on line 28
ANGLE : 0.947 15.272 20050
Ignored bad PDB record found on line 29
CHIRALITY : 0.057 0.244 2230
16 messages similar to the above omitted
Chain information for wt_model.pdb #1
---
Chain | Description
A B C D E | No description available
> coulombic
The following residues are missing heavy (non-hydrogen) atoms, which may
result in inaccurate electrostatics:
/A GLN 41
/A MET 47
/A GLN 49
/A LYS 50
/A ASP 51
/A PHE 375
/B GLN 41
/B MET 47
/B GLN 49
/B LYS 50
/B ASP 51
/B PHE 375
/C GLN 41
/C MET 47
/C GLN 49
/C LYS 50
/C ASP 51
/C PHE 375
/D GLN 41
/D MET 47
/D GLN 49
/D LYS 50
/D ASP 51
/D PHE 375
/E GLN 41
/E MET 47
/E GLN 49
/E LYS 50
/E ASP 51
/E PHE 375
Using Amber 20 recommended default charges and atom types for standard
residues
Coulombic values for wt_model.pdb_A SES surface #1.1: minimum, -17.47, mean
-2.40, maximum 12.76
Coulombic values for wt_model.pdb_B SES surface #1.2: minimum, -17.27, mean
-2.41, maximum 12.49
Coulombic values for wt_model.pdb_C SES surface #1.3: minimum, -17.02, mean
-2.42, maximum 12.37
Coulombic values for wt_model.pdb_D SES surface #1.4: minimum, -17.84, mean
-2.43, maximum 12.57
Coulombic values for wt_model.pdb_E SES surface #1.5: minimum, -17.67, mean
-2.42, maximum 13.30
To also show corresponding color key, enter the above coulombic command and
add key true
> close
> open 3mfp format mmcif fromDatabase pdb
3mfp title:
Atomic model of F-actin based on a 6.6 angstrom resolution cryoEM map [more
info...]
Chain information for 3mfp #1
---
Chain | Description
A | Actin, α skeletal muscle
Non-standard residues in 3mfp #1
---
ADP — adenosine-5'-diphosphate
3mfp mmCIF Assemblies
---
1| representative helical assembly
2| helical asymmetric unit
3| helical asymmetric unit, std helical frame
> coulombic
Using Amber 20 recommended default charges and atom types for standard
residues
Assigning partial charges to residue HIC (net charge +0) with am1-bcc method
Running ANTECHAMBER command: C:/Program Files/ChimeraX
1.2.5/bin/amber20/bin/antechamber -ek qm_theory='AM1', -i
C:\Users\myHP\AppData\Local\Temp\tmp4l_jhnpw\ante.in.mol2 -fi mol2 -o
C:\Users\myHP\AppData\Local\Temp\tmp4l_jhnpw\ante.out.mol2 -fo mol2 -c bcc -nc
0 -j 5 -s 2 -dr n
(HIC) ``
(HIC) `Welcome to antechamber 20.0: molecular input file processor.`
(HIC) ``
(HIC) `Info: Finished reading file
(C:\Users\myHP\AppData\Local\Temp\tmp4l_jhnpw\ante.in.mol2); atoms read (32),
bonds read (32).`
(HIC) `Info: Determining atomic numbers from atomic symbols which are case
sensitive.`
(HIC) `Running: C:/Program Files/ChimeraX 1.2.5/bin/amber20/bin/bondtype -j
part -i ANTECHAMBER_BOND_TYPE.AC0 -o ANTECHAMBER_BOND_TYPE.AC -f ac`
(HIC) `/usr/bin/antechamber: Fatal Error!`
(HIC) `Cannot properly run "C:/Program Files/ChimeraX
1.2.5/bin/amber20/bin/bondtype -j part -i ANTECHAMBER_BOND_TYPE.AC0 -o
ANTECHAMBER_BOND_TYPE.AC -f ac".`
(HIC) `No such file or directory`
Failure running ANTECHAMBER for residue HIC Check reply log for details
> select hic
Expected an objects specifier or a keyword
> help help:user
> help help:user
> close
> open 3d6y
Summary of feedback from opening 3d6y fetched from pdb
---
notes | Fetching compressed mmCIF 3d6y from
http://files.rcsb.org/download/3d6y.cif
Fetching CCD BER from http://ligand-expo.rcsb.org/reports/B/BER/BER.cif
Fetching CCD GOL from http://ligand-expo.rcsb.org/reports/G/GOL/GOL.cif
3d6y title:
Crystal structure of R275E mutant of BMRR bound to DNA and berberine [more
info...]
Chain information for 3d6y #1
---
Chain | Description
A | Multidrug-efflux transporter 1 regulator
B | BMR promoter DNA
Non-standard residues in 3d6y #1
---
BER — berberine
GOL — glycerol (glycerin; propane-1,2,3-triol)
3d6y mmCIF Assemblies
---
1| author_and_software_defined_assembly
> select :BER
25 atoms, 29 bonds, 1 residue, 1 model selected
> select :GOL
12 atoms, 10 bonds, 2 residues, 1 model selected
> select :GOL
12 atoms, 10 bonds, 2 residues, 1 model selected
> select clear
> select :GOL
12 atoms, 10 bonds, 2 residues, 1 model selected
> select :BER
25 atoms, 29 bonds, 1 residue, 1 model selected
> select :GOL
12 atoms, 10 bonds, 2 residues, 1 model selected
> close
> open 3d6y fromDatabase rcsb_bio maxAssemblies 1
Summary of feedback from opening 3d6y fetched from rcsb_bio
---
note | Fetching compressed 3d6y bioassembly 1 from
https://files.rcsb.org/download/3d6y.pdb1.gz
subunit 1 title:
Crystal structure of R275E mutant of BMRR bound to DNA and berberine [more
info...]
Chain information for subunit 1 #1.1
---
Chain | Description
A | No description available
B | No description available
Non-standard residues in subunit 1 #1.1
---
BER — berberine
GOL — glycerol (glycerin; propane-1,2,3-triol)
Chain information for subunit 2 #1.2
---
Chain | Description
A | No description available
B | No description available
Opened 1 biological assemblies for 3d6y
> hide #1.2 models
> show #1.2 models
> hide #1.1 models
> show #1.1 models
> hide #1.2 models
> show #1.2 models
> select :GOL
24 atoms, 20 bonds, 4 residues, 2 models selected
> select :GOL
24 atoms, 20 bonds, 4 residues, 2 models selected
> select :GOL
24 atoms, 20 bonds, 4 residues, 2 models selected
> select :BER
50 atoms, 58 bonds, 2 residues, 2 models selected
> select :GOL
24 atoms, 20 bonds, 4 residues, 2 models selected
> select :BER
50 atoms, 58 bonds, 2 residues, 2 models selected
> hide :gol
> hide solvent
> hide protein
> ~hide protein
Unknown command: ~hide protein
> show protein
> preset "initial styles" "original look"
Preset implemented in Python; no expansion to individual ChimeraX commands
available.
> hide protein
> preset "initial styles" "original look"
Preset implemented in Python; no expansion to individual ChimeraX commands
available.
> select clear
> hide protein
> preset "initial styles" "original look"
Preset implemented in Python; no expansion to individual ChimeraX commands
available.
> hide protein
> select clear
> coulombic
Using Amber 20 recommended default charges and atom types for standard
residues
Using Amber 20 recommended default charges and atom types for standard
residues
Coulombic values for subunit 1_A SES surface #1.1.1: minimum, -16.84, mean
-2.11, maximum 14.59
Coulombic values for subunit 1_B SES surface #1.1.2: minimum, -18.71, mean
-10.15, maximum -1.37
Coulombic values for subunit 2_A SES surface #1.2.1: minimum, -16.09, mean
-1.98, maximum 15.15
Coulombic values for subunit 2_B SES surface #1.2.2: minimum, -18.78, mean
-10.16, maximum -1.36
To also show corresponding color key, enter the above coulombic command and
add key true
> preset "initial styles" "original look"
Preset implemented in Python; no expansion to individual ChimeraX commands
available.
> hide :gol
> hide solvent
> hide protein
> coulombic protein range -10,10
Coulombic values for subunit 1_A SES surface #1.1.1: minimum, -16.84, mean
-2.11, maximum 14.59
Coulombic values for subunit 2_A SES surface #1.2.1: minimum, -16.09, mean
-1.98, maximum 15.15
> close
> open 3mfp format mmcif fromDatabase pdb
3mfp title:
Atomic model of F-actin based on a 6.6 angstrom resolution cryoEM map [more
info...]
Chain information for 3mfp #1
---
Chain | Description
A | Actin, α skeletal muscle
Non-standard residues in 3mfp #1
---
ADP — adenosine-5'-diphosphate
3mfp mmCIF Assemblies
---
1| representative helical assembly
2| helical asymmetric unit
3| helical asymmetric unit, std helical frame
> hide :ADP
> hide solvent
> hide protein
> select clear
> coulombic protein range -10,10
Using Amber 20 recommended default charges and atom types for standard
residues
Assigning partial charges to residue HIC (net charge +0) with am1-bcc method
Running ANTECHAMBER command: C:/Program Files/ChimeraX
1.2.5/bin/amber20/bin/antechamber -ek qm_theory='AM1', -i
C:\Users\myHP\AppData\Local\Temp\tmptnc6ju7l\ante.in.mol2 -fi mol2 -o
C:\Users\myHP\AppData\Local\Temp\tmptnc6ju7l\ante.out.mol2 -fo mol2 -c bcc -nc
0 -j 5 -s 2 -dr n
(HIC) ``
(HIC) `Welcome to antechamber 20.0: molecular input file processor.`
(HIC) ``
(HIC) `Info: Finished reading file
(C:\Users\myHP\AppData\Local\Temp\tmptnc6ju7l\ante.in.mol2); atoms read (32),
bonds read (32).`
(HIC) `Info: Determining atomic numbers from atomic symbols which are case
sensitive.`
(HIC) `Running: C:/Program Files/ChimeraX 1.2.5/bin/amber20/bin/bondtype -j
part -i ANTECHAMBER_BOND_TYPE.AC0 -o ANTECHAMBER_BOND_TYPE.AC -f ac`
(HIC) `/usr/bin/antechamber: Fatal Error!`
(HIC) `Cannot properly run "C:/Program Files/ChimeraX
1.2.5/bin/amber20/bin/bondtype -j part -i ANTECHAMBER_BOND_TYPE.AC0 -o
ANTECHAMBER_BOND_TYPE.AC -f ac".`
(HIC) `No such file or directory`
Failure running ANTECHAMBER for residue HIC Check reply log for details
> select :hic
11 atoms, 11 bonds, 1 residue, 1 model selected
Alignment identifier is 1/A
> select :his
80 atoms, 81 bonds, 8 residues, 1 model selected
> select clear
> select clear
> select :his
80 atoms, 81 bonds, 8 residues, 1 model selected
> ui tool show "Build Structure"
> ui tool show Updates
> toolshed show
> help help:devel
> help help:index.html
> rename #1 mutant4
> build modify :hic name his
Missing or invalid "atom" argument: Must specify exactly one atom (specified
11)
> select clear
> select :hic
11 atoms, 11 bonds, 1 residue, 1 model selected
> swapaa sel :his
Using Dunbrack library
/A HIC 73: phi -109.1, psi 24.1 trans
Dunbrack rotamer library does not support :HIS
> ui tool show "Command Line Interface"
> ui tool show Log
> select :h
Nothing selected
> select :his
80 atoms, 81 bonds, 8 residues, 1 model selected
> select :hic
11 atoms, 11 bonds, 1 residue, 1 model selected
> style sel ball
Changed 11 atom styles
> show sel
> color sel byhetero
> select clear
> coulombic
Using Amber 20 recommended default charges and atom types for standard
residues
Assigning partial charges to residue HIC (net charge +0) with am1-bcc method
Running ANTECHAMBER command: C:/Program Files/ChimeraX
1.2.5/bin/amber20/bin/antechamber -ek qm_theory='AM1', -i
C:\Users\myHP\AppData\Local\Temp\tmpbo0puy45\ante.in.mol2 -fi mol2 -o
C:\Users\myHP\AppData\Local\Temp\tmpbo0puy45\ante.out.mol2 -fo mol2 -c bcc -nc
0 -j 5 -s 2 -dr n
(HIC) ``
(HIC) `Welcome to antechamber 20.0: molecular input file processor.`
(HIC) ``
(HIC) `Info: Finished reading file
(C:\Users\myHP\AppData\Local\Temp\tmpbo0puy45\ante.in.mol2); atoms read (32),
bonds read (32).`
(HIC) `Info: Determining atomic numbers from atomic symbols which are case
sensitive.`
(HIC) `Running: C:/Program Files/ChimeraX 1.2.5/bin/amber20/bin/bondtype -j
part -i ANTECHAMBER_BOND_TYPE.AC0 -o ANTECHAMBER_BOND_TYPE.AC -f ac`
(HIC) `/usr/bin/antechamber: Fatal Error!`
(HIC) `Cannot properly run "C:/Program Files/ChimeraX
1.2.5/bin/amber20/bin/bondtype -j part -i ANTECHAMBER_BOND_TYPE.AC0 -o
ANTECHAMBER_BOND_TYPE.AC -f ac".`
(HIC) `No such file or directory`
Failure running ANTECHAMBER for residue HIC Check reply log for details
> select hic
Expected an objects specifier or a keyword
> select :hic
11 atoms, 11 bonds, 1 residue, 1 model selected
> coulombic :hic range -10,10
Using Amber 20 recommended default charges and atom types for standard
residues
Assigning partial charges to residue HIC (net charge +0) with am1-bcc method
Running ANTECHAMBER command: C:/Program Files/ChimeraX
1.2.5/bin/amber20/bin/antechamber -ek qm_theory='AM1', -i
C:\Users\myHP\AppData\Local\Temp\tmpxvxquhmd\ante.in.mol2 -fi mol2 -o
C:\Users\myHP\AppData\Local\Temp\tmpxvxquhmd\ante.out.mol2 -fo mol2 -c bcc -nc
0 -j 5 -s 2 -dr n
(HIC) ``
(HIC) `Welcome to antechamber 20.0: molecular input file processor.`
(HIC) ``
(HIC) `Info: Finished reading file
(C:\Users\myHP\AppData\Local\Temp\tmpxvxquhmd\ante.in.mol2); atoms read (32),
bonds read (32).`
(HIC) `Info: Determining atomic numbers from atomic symbols which are case
sensitive.`
(HIC) `Running: C:/Program Files/ChimeraX 1.2.5/bin/amber20/bin/bondtype -j
part -i ANTECHAMBER_BOND_TYPE.AC0 -o ANTECHAMBER_BOND_TYPE.AC -f ac`
(HIC) `/usr/bin/antechamber: Fatal Error!`
(HIC) `Cannot properly run "C:/Program Files/ChimeraX
1.2.5/bin/amber20/bin/bondtype -j part -i ANTECHAMBER_BOND_TYPE.AC0 -o
ANTECHAMBER_BOND_TYPE.AC -f ac".`
(HIC) `No such file or directory`
Failure running ANTECHAMBER for residue HIC Check reply log for details
> hide sel
> select clear
> coulombic protein range -10,10
Using Amber 20 recommended default charges and atom types for standard
residues
Assigning partial charges to residue HIC (net charge +0) with am1-bcc method
Running ANTECHAMBER command: C:/Program Files/ChimeraX
1.2.5/bin/amber20/bin/antechamber -ek qm_theory='AM1', -i
C:\Users\myHP\AppData\Local\Temp\tmp8t49fojj\ante.in.mol2 -fi mol2 -o
C:\Users\myHP\AppData\Local\Temp\tmp8t49fojj\ante.out.mol2 -fo mol2 -c bcc -nc
0 -j 5 -s 2 -dr n
(HIC) ``
(HIC) `Welcome to antechamber 20.0: molecular input file processor.`
(HIC) ``
(HIC) `Info: Finished reading file
(C:\Users\myHP\AppData\Local\Temp\tmp8t49fojj\ante.in.mol2); atoms read (32),
bonds read (32).`
(HIC) `Info: Determining atomic numbers from atomic symbols which are case
sensitive.`
(HIC) `Running: C:/Program Files/ChimeraX 1.2.5/bin/amber20/bin/bondtype -j
part -i ANTECHAMBER_BOND_TYPE.AC0 -o ANTECHAMBER_BOND_TYPE.AC -f ac`
(HIC) `/usr/bin/antechamber: Fatal Error!`
(HIC) `Cannot properly run "C:/Program Files/ChimeraX
1.2.5/bin/amber20/bin/bondtype -j part -i ANTECHAMBER_BOND_TYPE.AC0 -o
ANTECHAMBER_BOND_TYPE.AC -f ac".`
(HIC) `No such file or directory`
Failure running ANTECHAMBER for residue HIC Check reply log for details
> coulombic protein range -10,10
Using Amber 20 recommended default charges and atom types for standard
residues
Assigning partial charges to residue HIC (net charge +0) with am1-bcc method
Running ANTECHAMBER command: C:/Program Files/ChimeraX
1.2.5/bin/amber20/bin/antechamber -ek qm_theory='AM1', -i
C:\Users\myHP\AppData\Local\Temp\tmp9teu92i_\ante.in.mol2 -fi mol2 -o
C:\Users\myHP\AppData\Local\Temp\tmp9teu92i_\ante.out.mol2 -fo mol2 -c bcc -nc
0 -j 5 -s 2 -dr n
(HIC) ``
(HIC) `Welcome to antechamber 20.0: molecular input file processor.`
(HIC) ``
(HIC) `Info: Finished reading file
(C:\Users\myHP\AppData\Local\Temp\tmp9teu92i_\ante.in.mol2); atoms read (32),
bonds read (32).`
(HIC) `Info: Determining atomic numbers from atomic symbols which are case
sensitive.`
(HIC) `Running: C:/Program Files/ChimeraX 1.2.5/bin/amber20/bin/bondtype -j
part -i ANTECHAMBER_BOND_TYPE.AC0 -o ANTECHAMBER_BOND_TYPE.AC -f ac`
(HIC) `/usr/bin/antechamber: Fatal Error!`
(HIC) `Cannot properly run "C:/Program Files/ChimeraX
1.2.5/bin/amber20/bin/bondtype -j part -i ANTECHAMBER_BOND_TYPE.AC0 -o
ANTECHAMBER_BOND_TYPE.AC -f ac".`
(HIC) `No such file or directory`
Failure running ANTECHAMBER for residue HIC Check reply log for details
> coulombic :1-72, 74-375 range -10,10
Using Amber 20 recommended default charges and atom types for standard
residues
Assigning partial charges to residue HIC (net charge +0) with am1-bcc method
Running ANTECHAMBER command: C:/Program Files/ChimeraX
1.2.5/bin/amber20/bin/antechamber -ek qm_theory='AM1', -i
C:\Users\myHP\AppData\Local\Temp\tmp53jgcy2f\ante.in.mol2 -fi mol2 -o
C:\Users\myHP\AppData\Local\Temp\tmp53jgcy2f\ante.out.mol2 -fo mol2 -c bcc -nc
0 -j 5 -s 2 -dr n
(HIC) ``
(HIC) `Welcome to antechamber 20.0: molecular input file processor.`
(HIC) ``
(HIC) `Info: Finished reading file
(C:\Users\myHP\AppData\Local\Temp\tmp53jgcy2f\ante.in.mol2); atoms read (32),
bonds read (32).`
(HIC) `Info: Determining atomic numbers from atomic symbols which are case
sensitive.`
(HIC) `Running: C:/Program Files/ChimeraX 1.2.5/bin/amber20/bin/bondtype -j
part -i ANTECHAMBER_BOND_TYPE.AC0 -o ANTECHAMBER_BOND_TYPE.AC -f ac`
(HIC) `/usr/bin/antechamber: Fatal Error!`
(HIC) `Cannot properly run "C:/Program Files/ChimeraX
1.2.5/bin/amber20/bin/bondtype -j part -i ANTECHAMBER_BOND_TYPE.AC0 -o
ANTECHAMBER_BOND_TYPE.AC -f ac".`
(HIC) `No such file or directory`
Failure running ANTECHAMBER for residue HIC Check reply log for details
> delete :hic
> coulombic protein range -10,10
Using Amber 20 recommended default charges and atom types for standard
residues
Coulombic values for mutant4_A SES surface #1.2: minimum, -18.10, mean -2.62,
maximum 10.75
> select :6110
Nothing selected
> select :61
9 atoms, 8 bonds, 1 residue, 1 model selected
> preset "initial styles" "original look"
Preset implemented in Python; no expansion to individual ChimeraX commands
available.
> close
> open 3mfp format mmcif fromDatabase pdb
3mfp title:
Atomic model of F-actin based on a 6.6 angstrom resolution cryoEM map [more
info...]
Chain information for 3mfp #1
---
Chain | Description
A | Actin, α skeletal muscle
Non-standard residues in 3mfp #1
---
ADP — adenosine-5'-diphosphate
3mfp mmCIF Assemblies
---
1| representative helical assembly
2| helical asymmetric unit
3| helical asymmetric unit, std helical frame
> color bynucleotide
> color bynucleotide
Alignment identifier is 1/A
> surface
> coulombic
Using Amber 20 recommended default charges and atom types for standard
residues
Assigning partial charges to residue HIC (net charge +0) with am1-bcc method
Running ANTECHAMBER command: C:/Program Files/ChimeraX
1.2.5/bin/amber20/bin/antechamber -ek qm_theory='AM1', -i
C:\Users\myHP\AppData\Local\Temp\tmphr9qv0v0\ante.in.mol2 -fi mol2 -o
C:\Users\myHP\AppData\Local\Temp\tmphr9qv0v0\ante.out.mol2 -fo mol2 -c bcc -nc
0 -j 5 -s 2 -dr n
(HIC) ``
(HIC) `Welcome to antechamber 20.0: molecular input file processor.`
(HIC) ``
(HIC) `Info: Finished reading file
(C:\Users\myHP\AppData\Local\Temp\tmphr9qv0v0\ante.in.mol2); atoms read (32),
bonds read (32).`
(HIC) `Info: Determining atomic numbers from atomic symbols which are case
sensitive.`
(HIC) `Running: C:/Program Files/ChimeraX 1.2.5/bin/amber20/bin/bondtype -j
part -i ANTECHAMBER_BOND_TYPE.AC0 -o ANTECHAMBER_BOND_TYPE.AC -f ac`
(HIC) `/usr/bin/antechamber: Fatal Error!`
(HIC) `Cannot properly run "C:/Program Files/ChimeraX
1.2.5/bin/amber20/bin/bondtype -j part -i ANTECHAMBER_BOND_TYPE.AC0 -o
ANTECHAMBER_BOND_TYPE.AC -f ac".`
(HIC) `No such file or directory`
Failure running ANTECHAMBER for residue HIC Check reply log for details
> preset "initial styles" "original look"
Preset implemented in Python; no expansion to individual ChimeraX commands
available.
OpenGL version: 3.3.14739 Core Profile Forward-Compatible Context 21.6.1 27.20.22001.14011
OpenGL renderer: AMD Radeon(TM) RX Vega 10 Graphics
OpenGL vendor: ATI Technologies Inc.
Manufacturer: HP
Model: HP Laptop 17-ca1xxx
OS: Microsoft Windows 10 Home (Build 19042)
Memory: 14,942,511,104
MaxProcessMemory: 137,438,953,344
CPU: 8 AMD Ryzen 7 3700U with Radeon Vega Mobile Gfx
OSLanguage: en-US
Locale: ('en_US', 'cp936')
PyQt5 5.15.2, Qt 5.15.2
Installed Packages:
alabaster: 0.7.12
appdirs: 1.4.4
Babel: 2.9.1
backcall: 0.2.0
blockdiag: 2.0.1
certifi: 2020.12.5
cftime: 1.5.0
chardet: 3.0.4
ChimeraX-AddCharge: 1.0.1
ChimeraX-AddH: 2.1.6
ChimeraX-AlignmentAlgorithms: 2.0
ChimeraX-AlignmentHdrs: 3.2
ChimeraX-AlignmentMatrices: 2.0
ChimeraX-Alignments: 2.1
ChimeraX-AmberInfo: 1.0
ChimeraX-Arrays: 1.0
ChimeraX-Atomic: 1.13.2
ChimeraX-AtomicLibrary: 3.1.3
ChimeraX-AtomSearch: 2.0
ChimeraX-AtomSearchLibrary: 1.0
ChimeraX-AxesPlanes: 2.0
ChimeraX-BasicActions: 1.1
ChimeraX-BILD: 1.0
ChimeraX-BlastProtein: 1.1
ChimeraX-BondRot: 2.0
ChimeraX-BugReporter: 1.0
ChimeraX-BuildStructure: 2.5.2
ChimeraX-Bumps: 1.0
ChimeraX-BundleBuilder: 1.1
ChimeraX-ButtonPanel: 1.0
ChimeraX-CageBuilder: 1.0
ChimeraX-CellPack: 1.0
ChimeraX-Centroids: 1.1
ChimeraX-ChemGroup: 2.0
ChimeraX-Clashes: 2.1
ChimeraX-ColorActions: 1.0
ChimeraX-ColorGlobe: 1.0
ChimeraX-ColorKey: 1.2.1
ChimeraX-CommandLine: 1.1.4
ChimeraX-ConnectStructure: 2.0
ChimeraX-Contacts: 1.0
ChimeraX-Core: 1.2.5
ChimeraX-CoreFormats: 1.0
ChimeraX-coulombic: 1.1.1
ChimeraX-Crosslinks: 1.0
ChimeraX-Crystal: 1.0
ChimeraX-CrystalContacts: 1.0
ChimeraX-DataFormats: 1.1
ChimeraX-Dicom: 1.0
ChimeraX-DistMonitor: 1.1.3
ChimeraX-DistUI: 1.0
ChimeraX-Dssp: 2.0
ChimeraX-EMDB-SFF: 1.0
ChimeraX-ExperimentalCommands: 1.0
ChimeraX-FileHistory: 1.0
ChimeraX-FunctionKey: 1.0
ChimeraX-Geometry: 1.1
ChimeraX-gltf: 1.0
ChimeraX-Graphics: 1.0
ChimeraX-Hbonds: 2.1
ChimeraX-Help: 1.1
ChimeraX-HKCage: 1.3
ChimeraX-IHM: 1.0
ChimeraX-ImageFormats: 1.1
ChimeraX-IMOD: 1.0
ChimeraX-IO: 1.0.1
ChimeraX-Label: 1.0
ChimeraX-ListInfo: 1.1.1
ChimeraX-Log: 1.1.2
ChimeraX-LookingGlass: 1.1
ChimeraX-Maestro: 1.8.1
ChimeraX-Map: 1.0.2
ChimeraX-MapData: 2.0
ChimeraX-MapEraser: 1.0
ChimeraX-MapFilter: 2.0
ChimeraX-MapFit: 2.0
ChimeraX-MapSeries: 2.0
ChimeraX-Markers: 1.0
ChimeraX-Mask: 1.0
ChimeraX-MatchMaker: 1.2.1
ChimeraX-MDcrds: 2.2
ChimeraX-MedicalToolbar: 1.0.1
ChimeraX-Meeting: 1.0
ChimeraX-MLP: 1.1
ChimeraX-mmCIF: 2.3
ChimeraX-MMTF: 2.1
ChimeraX-Modeller: 1.0.1
ChimeraX-ModelPanel: 1.0.1
ChimeraX-ModelSeries: 1.0
ChimeraX-Mol2: 2.0
ChimeraX-Morph: 1.0
ChimeraX-MouseModes: 1.1
ChimeraX-Movie: 1.0
ChimeraX-Neuron: 1.0
ChimeraX-Nucleotides: 2.0.1
ChimeraX-OpenCommand: 1.5
ChimeraX-PDB: 2.4.1
ChimeraX-PDBBio: 1.0
ChimeraX-PDBLibrary: 1.0.1
ChimeraX-PDBMatrices: 1.0
ChimeraX-PickBlobs: 1.0
ChimeraX-Positions: 1.0
ChimeraX-PresetMgr: 1.0.1
ChimeraX-PubChem: 2.0.1
ChimeraX-ReadPbonds: 1.0
ChimeraX-Registration: 1.1
ChimeraX-RemoteControl: 1.0
ChimeraX-ResidueFit: 1.0
ChimeraX-RestServer: 1.1
ChimeraX-RNALayout: 1.0
ChimeraX-RotamerLibMgr: 2.0
ChimeraX-RotamerLibsDunbrack: 2.0
ChimeraX-RotamerLibsDynameomics: 2.0
ChimeraX-RotamerLibsRichardson: 2.0
ChimeraX-SaveCommand: 1.4
ChimeraX-SchemeMgr: 1.0
ChimeraX-SDF: 2.0
ChimeraX-Segger: 1.0
ChimeraX-Segment: 1.0
ChimeraX-SeqView: 2.3
ChimeraX-Shape: 1.0.1
ChimeraX-Shell: 1.0
ChimeraX-Shortcuts: 1.0
ChimeraX-ShowAttr: 1.0
ChimeraX-ShowSequences: 1.0
ChimeraX-SideView: 1.0
ChimeraX-Smiles: 2.0.1
ChimeraX-SmoothLines: 1.0
ChimeraX-SpaceNavigator: 1.0
ChimeraX-StdCommands: 1.3.1
ChimeraX-STL: 1.0
ChimeraX-Storm: 1.0
ChimeraX-Struts: 1.0
ChimeraX-Surface: 1.0
ChimeraX-SwapAA: 2.0
ChimeraX-SwapRes: 2.1
ChimeraX-TapeMeasure: 1.0
ChimeraX-Test: 1.0
ChimeraX-Toolbar: 1.0.1
ChimeraX-ToolshedUtils: 1.2
ChimeraX-Tug: 1.0
ChimeraX-UI: 1.7.6
ChimeraX-uniprot: 2.1
ChimeraX-UnitCell: 1.0
ChimeraX-ViewDockX: 1.0
ChimeraX-Vive: 1.1
ChimeraX-VolumeMenu: 1.0
ChimeraX-VTK: 1.0
ChimeraX-WavefrontOBJ: 1.0
ChimeraX-WebCam: 1.0
ChimeraX-WebServices: 1.0
ChimeraX-Zone: 1.0
colorama: 0.4.3
comtypes: 1.1.7
cxservices: 1.0
cycler: 0.10.0
Cython: 0.29.21
decorator: 5.0.9
distlib: 0.3.1
docutils: 0.16
filelock: 3.0.12
funcparserlib: 0.3.6
grako: 3.16.5
h5py: 2.10.0
html2text: 2020.1.16
idna: 2.10
ihm: 0.17
imagecodecs: 2020.5.30
imagesize: 1.2.0
ipykernel: 5.3.4
ipython: 7.18.1
ipython-genutils: 0.2.0
jedi: 0.17.2
Jinja2: 2.11.2
jupyter-client: 6.1.7
jupyter-core: 4.7.1
kiwisolver: 1.3.1
line-profiler: 2.1.2
lxml: 4.6.2
lz4: 3.1.0
MarkupSafe: 2.0.1
matplotlib: 3.3.2
msgpack: 1.0.0
netCDF4: 1.5.4
networkx: 2.5
numexpr: 2.7.3
numpy: 1.19.2
numpydoc: 1.1.0
openvr: 1.14.1501
packaging: 20.9
ParmEd: 3.2.0
parso: 0.7.1
pickleshare: 0.7.5
Pillow: 7.2.0
pip: 21.0.1
pkginfo: 1.5.0.1
prompt-toolkit: 3.0.18
psutil: 5.7.2
pycollada: 0.7.1
pydicom: 2.0.0
Pygments: 2.7.1
PyOpenGL: 3.1.5
PyOpenGL-accelerate: 3.1.5
pyparsing: 2.4.7
PyQt5-commercial: 5.15.2
PyQt5-sip: 12.8.1
PyQtWebEngine-commercial: 5.15.2
python-dateutil: 2.8.1
pytz: 2021.1
pywin32: 228
pyzmq: 22.0.3
qtconsole: 4.7.7
QtPy: 1.9.0
RandomWords: 0.3.0
requests: 2.24.0
scipy: 1.5.2
setuptools: 50.3.2
sfftk-rw: 0.6.7.dev1
six: 1.15.0
snowballstemmer: 2.1.0
sortedcontainers: 2.2.2
Sphinx: 3.2.1
sphinxcontrib-applehelp: 1.0.2
sphinxcontrib-blockdiag: 2.0.0
sphinxcontrib-devhelp: 1.0.2
sphinxcontrib-htmlhelp: 2.0.0
sphinxcontrib-jsmath: 1.0.1
sphinxcontrib-qthelp: 1.0.3
sphinxcontrib-serializinghtml: 1.1.5
suds-jurko: 0.6
tables: 3.6.1
tifffile: 2020.9.3
tinyarray: 1.2.3
tornado: 6.1
traitlets: 5.0.5
urllib3: 1.25.11
wcwidth: 0.2.5
webcolors: 1.11.1
wheel: 0.36.0
wheel-filename: 1.3.0
WMI: 1.5.1
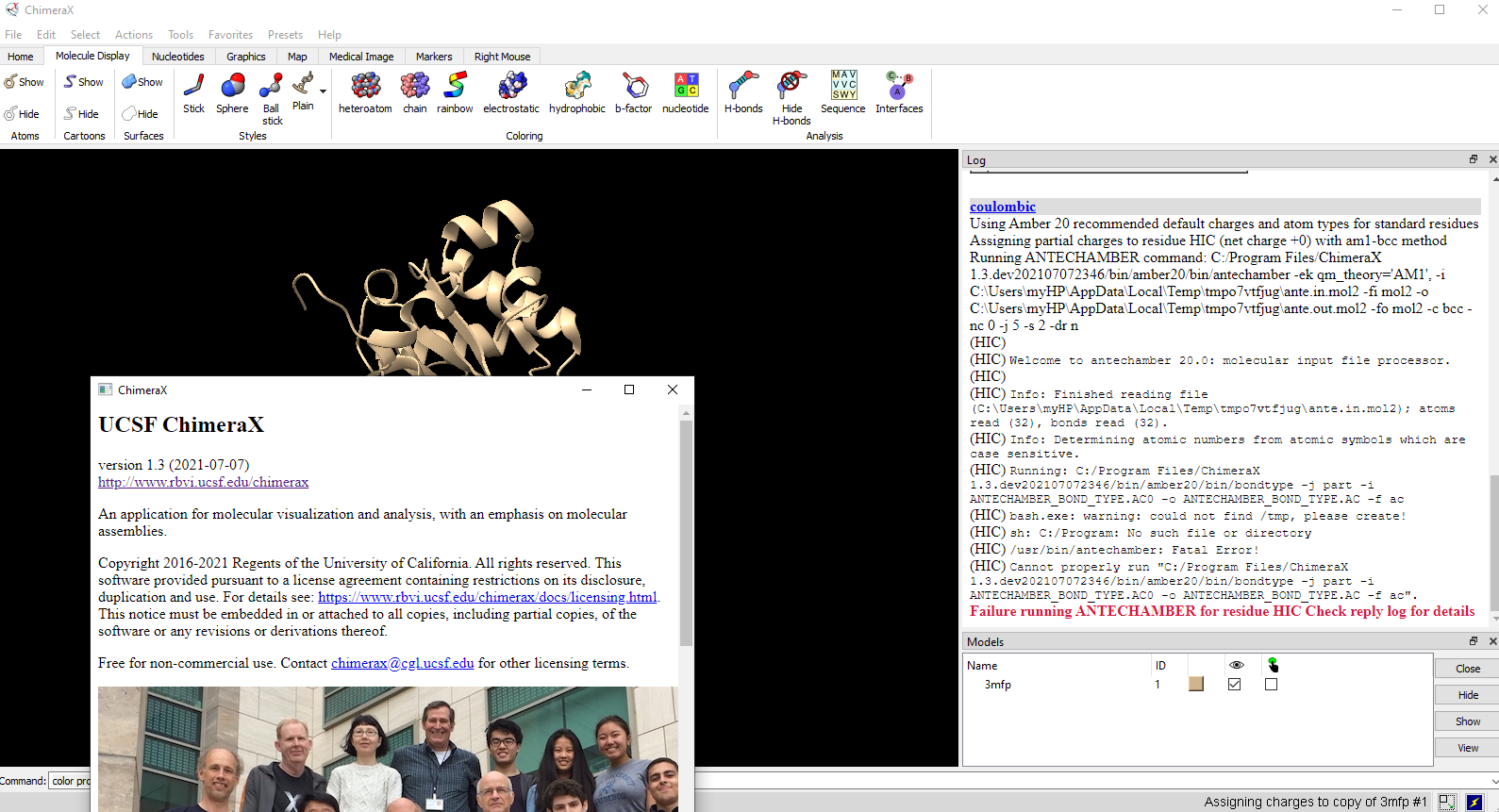
File attachment: antechanber error.PNG
{kind=link}
{kind=link}
Attachments (2)
{kind=link}
{kind=link}
Change History (15)
by , 5 years ago
| Attachment: | antechanber error.PNG added |
|---|
comment:1 by , 5 years ago
| Cc: | added |
|---|---|
| Component: | Unassigned → Structure Editing |
| Owner: | set to |
| Platform: | → all |
| Project: | → ChimeraX |
| Status: | new → accepted |
| Summary: | ChimeraX bug report submission → Failure running antechamber on Windows |
comment:2 by , 5 years ago
| Resolution: | → fixed |
|---|---|
| Status: | accepted → closed |
Should be fixed in tomorrow's build. Thanks for reporting this problem.
comment:3 by , 5 years ago
Thanks! Though the fix has not in windows daily version yet, chimerax-daily.exe<https://www.rbvi.ucsf.edu/chimerax/cgi-bin/secure/chimerax-get.py?file=windows/chimerax-daily.exe>.
Best regards!
Wenxiang
From: ChimeraX<mailto:ChimeraX-bugs-admin@cgl.ucsf.edu>
Sent: Tuesday, July 6, 2021 8:44 PM
Cc: chimera-programmers@cgl.ucsf.edu<mailto:chimera-programmers@cgl.ucsf.edu>; pett@cgl.ucsf.edu<mailto:pett@cgl.ucsf.edu>; Cao, Wenxiang<mailto:wenxiang.cao@yale.edu>
Subject: Re: [ChimeraX] #4861: Failure running antechamber on Windows
#4861: Failure running antechamber on Windows
----------------------------------------+----------------------------
Reporter: wenxiang.cao@… | Owner: Eric Pettersen
Type: defect | Status: closed
Priority: normal | Milestone:
Component: Structure Editing | Version:
Resolution: fixed | Keywords:
Blocked By: | Blocking:
Notify when closed: | Platform: all
Project: ChimeraX |
----------------------------------------+----------------------------
Changes (by Eric Pettersen):
* status: accepted => closed
* resolution: => fixed
Comment:
Should be fixed in tomorrow's build. Thanks for reporting this problem.
--
Ticket URL: <https://nam12.safelinks.protection.outlook.com/?url=https%3A%2F%2Fplato.cgl.ucsf.edu%2Ftrac%2FChimeraX%2Fticket%2F4861%23comment%3A2&data=04%7C01%7Cwenxiang.cao%40yale.edu%7C15c50ec379294dde8a9208d940e0526f%7Cdd8cbebb21394df8b4114e3e87abeb5c%7C0%7C0%7C637612154480158788%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C1000&sdata=6HyL4a8CS9GGjh5t7dQoLVWdl0Oya5eU%2FE8MbvXeQyY%3D&reserved=0>
ChimeraX <https://nam12.safelinks.protection.outlook.com/?url=http%3A%2F%2Fwww.rbvi.ucsf.edu%2Fchimerax%2F&data=04%7C01%7Cwenxiang.cao%40yale.edu%7C15c50ec379294dde8a9208d940e0526f%7Cdd8cbebb21394df8b4114e3e87abeb5c%7C0%7C0%7C637612154480158788%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C1000&sdata=K46Ye20lFABuJ1Vsj9aQf%2BihV9PNZUwFDS%2FBf5KBu9A%3D&reserved=0>
ChimeraX Issue Tracker
follow-up: 3 comment:4 by , 5 years ago
| Resolution: | fixed |
|---|---|
| Status: | closed → reopened |
You are right -- the fix isn't in the build. I'll re-close this ticket when I am sure the fix is available.
--Eric
comment:5 by , 5 years ago
Thank you Eric!
Wenxiang
From: ChimeraX<mailto:ChimeraX-bugs-admin@cgl.ucsf.edu>
Sent: Wednesday, July 7, 2021 4:53 PM
Cc: chimera-programmers@cgl.ucsf.edu<mailto:chimera-programmers@cgl.ucsf.edu>; pett@cgl.ucsf.edu<mailto:pett@cgl.ucsf.edu>; Cao, Wenxiang<mailto:wenxiang.cao@yale.edu>
Subject: Re: [ChimeraX] #4861: Failure running antechamber on Windows
#4861: Failure running antechamber on Windows
----------------------------------------+----------------------------
Reporter: wenxiang.cao@… | Owner: Eric Pettersen
Type: defect | Status: reopened
Priority: normal | Milestone:
Component: Structure Editing | Version:
Resolution: | Keywords:
Blocked By: | Blocking:
Notify when closed: | Platform: all
Project: ChimeraX |
----------------------------------------+----------------------------
Changes (by Eric Pettersen):
* status: closed => reopened
* resolution: fixed =>
Comment:
You are right -- the fix isn't in the build. I'll re-close this ticket
when I am sure the fix is available.
--Eric
--
Ticket URL: <https://nam12.safelinks.protection.outlook.com/?url=https%3A%2F%2Fwww.rbvi.ucsf.edu%2Ftrac%2FChimeraX%2Fticket%2F4861%23comment%3A4&data=04%7C01%7Cwenxiang.cao%40yale.edu%7Cdf2421a2ee0a4fd94e5d08d9418942ef%7Cdd8cbebb21394df8b4114e3e87abeb5c%7C0%7C0%7C637612880052005268%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C1000&sdata=rJvaBIdLK2M4eZOqpY%2BnPDBYTAPfZaYfgCiaonQ0QR0%3D&reserved=0>
ChimeraX <https://nam12.safelinks.protection.outlook.com/?url=https%3A%2F%2Fwww.rbvi.ucsf.edu%2Fchimerax%2F&data=04%7C01%7Cwenxiang.cao%40yale.edu%7Cdf2421a2ee0a4fd94e5d08d9418942ef%7Cdd8cbebb21394df8b4114e3e87abeb5c%7C0%7C0%7C637612880052005268%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C1000&sdata=LaKplKXkBSfYzN%2FTsoQI%2FZlOObTuc75IB%2Buu75VjFog%3D&reserved=0>
ChimeraX Issue Tracker
follow-up: 5 comment:6 by , 5 years ago
Last night's Windows build failed (code signing problem). The date when the build was made is on the download page.
comment:8 by , 5 years ago
Dear Eric:
Just for your information that the bug is still in the 21-07-07 version. Please see the attached screen shot.
Thanks!
Wenxiang
From: ChimeraX<mailto:ChimeraX-bugs-admin@cgl.ucsf.edu>
Sent: Wednesday, July 7, 2021 5:21 PM
Cc: chimera-programmers@cgl.ucsf.edu<mailto:chimera-programmers@cgl.ucsf.edu>; pett@cgl.ucsf.edu<mailto:pett@cgl.ucsf.edu>; Cao, Wenxiang<mailto:wenxiang.cao@yale.edu>
Subject: Re: [ChimeraX] #4861: Failure running antechamber on Windows
#4861: Failure running antechamber on Windows
----------------------------------------+----------------------------
Reporter: wenxiang.cao@… | Owner: Eric Pettersen
Type: defect | Status: reopened
Priority: normal | Milestone:
Component: Structure Editing | Version:
Resolution: | Keywords:
Blocked By: | Blocking:
Notify when closed: | Platform: all
Project: ChimeraX |
----------------------------------------+----------------------------
Comment (by Eric Pettersen):
Ah, right!
--
Ticket URL: <https://nam12.safelinks.protection.outlook.com/?url=https%3A%2F%2Fwww.rbvi.ucsf.edu%2Ftrac%2FChimeraX%2Fticket%2F4861%23comment%3A7&data=04%7C01%7Cwenxiang.cao%40yale.edu%7Cb8209408aa944df0e54608d9418d25ae%7Cdd8cbebb21394df8b4114e3e87abeb5c%7C0%7C1%7C637612896766906176%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000&sdata=qkzNkehLnwbuENLQfYfrfYSo6SXca%2B8vcRSXk3Xf85Q%3D&reserved=0>
ChimeraX <https://nam12.safelinks.protection.outlook.com/?url=https%3A%2F%2Fwww.rbvi.ucsf.edu%2Fchimerax%2F&data=04%7C01%7Cwenxiang.cao%40yale.edu%7Cb8209408aa944df0e54608d9418d25ae%7Cdd8cbebb21394df8b4114e3e87abeb5c%7C0%7C1%7C637612896766906176%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C3000&sdata=qSc4h2Cm7Zql%2FdAo2Wuc2y81A0QPIdstc0PDtMMkMlk%3D&reserved=0>
ChimeraX Issue Tracker
follow-up: 8 comment:9 by , 5 years ago
The cause of the problem is that AmberTools had a code regression where they no longer properly quote executable paths that have embedded spaces. I will have to look at the very latest AmberTools source to verify that the regression is still there, and then interact with the AmberTools developers to get them to rectify the problem if it is. In the interim, I will have to patch our own AmberTools code to work around the problem.
comment:11 by , 5 years ago
| Resolution: | → fixed |
|---|---|
| Status: | reopened → closed |
Pretty sure the problem is fixed now (same problem on Mac is fixed). Let me know if it isn't.
comment:12 by , 5 years ago
Dear Eric:
The problem is fixed in Windows 10.
Thanks a lot!
Wenxiang
From: ChimeraX<mailto:ChimeraX-bugs-admin@cgl.ucsf.edu>
Sent: Wednesday, July 14, 2021 12:24 PM
Cc: chimera-programmers@cgl.ucsf.edu<mailto:chimera-programmers@cgl.ucsf.edu>; pett@cgl.ucsf.edu<mailto:pett@cgl.ucsf.edu>; Cao, Wenxiang<mailto:wenxiang.cao@yale.edu>
Subject: Re: [ChimeraX] #4861: Failure running antechamber on Windows
#4861: Failure running antechamber on Windows
----------------------------------------+----------------------------
Reporter: wenxiang.cao@… | Owner: Eric Pettersen
Type: defect | Status: closed
Priority: normal | Milestone:
Component: Structure Editing | Version:
Resolution: fixed | Keywords:
Blocked By: | Blocking:
Notify when closed: | Platform: all
Project: ChimeraX |
----------------------------------------+----------------------------
Changes (by Eric Pettersen):
* status: reopened => closed
* resolution: => fixed
Comment:
Pretty sure the problem is fixed now (same problem on Mac is fixed). Let
me know if it isn't.
--
Ticket URL: <https://nam12.safelinks.protection.outlook.com/?url=https%3A%2F%2Fwww.rbvi.ucsf.edu%2Ftrac%2FChimeraX%2Fticket%2F4861%23comment%3A11&data=04%7C01%7Cwenxiang.cao%40yale.edu%7Ceba02a64b0fa43aebf6308d946e3e4fe%7Cdd8cbebb21394df8b4114e3e87abeb5c%7C0%7C0%7C637618766886087557%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C1000&sdata=h93VkEXEWlSbVuKrGh8wp%2BL9yJDxYUQDyITzpefjaos%3D&reserved=0>
ChimeraX <https://nam12.safelinks.protection.outlook.com/?url=https%3A%2F%2Fwww.rbvi.ucsf.edu%2Fchimerax%2F&data=04%7C01%7Cwenxiang.cao%40yale.edu%7Ceba02a64b0fa43aebf6308d946e3e4fe%7Cdd8cbebb21394df8b4114e3e87abeb5c%7C0%7C0%7C637618766886097551%7CUnknown%7CTWFpbGZsb3d8eyJWIjoiMC4wLjAwMDAiLCJQIjoiV2luMzIiLCJBTiI6Ik1haWwiLCJXVCI6Mn0%3D%7C1000&sdata=g2Q9EqFM0HQRtDbvqG1OjMCNQLx%2BxsCEvNA67osR9yw%3D&reserved=0>
ChimeraX Issue Tracker
Note:
See TracTickets
for help on using tickets.
Added by email2trac