Opened 6 years ago
Closed 3 years ago
#3407 closed enhancement (worksforme)
Maximum common substructure
| Reported by: | Tristan Croll | Owned by: | Tristan Croll |
|---|---|---|---|
| Priority: | normal | Milestone: | |
| Component: | Structure Comparison | Version: | |
| Keywords: | Cc: | Eric Pettersen, Goddard | |
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
The following bug report has been submitted: Platform: Linux-3.10.0-1062.9.1.el7.x86_64-x86_64-with-centos-7.7.1908-Core ChimeraX Version: 1.0 (2020-06-04 23:15:07 UTC) Description There are a few places where it would be really nice to have a fast algorithm available to find the maximum common substructure between two residues: - suggesting MD templates to match a given residue in a residue- and atom-name agnostic manner (and then adding missing atoms and removing superfluous ones) - "mutating" one residue to another while maintaining the maximum possible number of existing atom positions (e.g. swapping lipid head-groups, or switching between related drug-like compounds) The challenge is that this is a hard, NP-complete problem. The algorithm available in `networkx.isomorphism.ISMAGS.largest_common_subgraph()` seems fast when matching by element as long as the second graph given in its __init__ is already a subgraph of the first - but as soon as the second has even a few superfluous atoms it slows to a crawl - e.g. taking many minutes to match phosphatidylcholine to phosphatidylethanolamine (I don't know exactly how long - I gave up waiting after about 10 minutes). RDKit has a fast algorithm implemented (https://www.rdkit.org/docs/GettingStartedInPython.html#maximum-common-substructure; https://github.com/rdkit/rdkit/tree/master/Code/GraphMol/FMCS) and is BSD-licensed with releases for all platforms on Conda... although not for Python 3.8 yet. Any thoughts? OpenGL version: 3.3.0 NVIDIA 440.33.01 OpenGL renderer: TITAN Xp/PCIe/SSE2 OpenGL vendor: NVIDIA Corporation Manufacturer: Dell Inc. Model: Precision T5600 OS: CentOS Linux 7 Core Architecture: 64bit ELF CPU: 32 Intel(R) Xeon(R) CPU E5-2687W 0 @ 3.10GHz Cache Size: 20480 KB Memory: total used free shared buff/cache available Mem: 62G 5.6G 43G 374M 13G 56G Swap: 4.9G 0B 4.9G Graphics: 03:00.0 VGA compatible controller [0300]: NVIDIA Corporation GP102 [TITAN Xp] [10de:1b02] (rev a1) Subsystem: NVIDIA Corporation Device [10de:11df] Kernel driver in use: nvidia PyQt version: 5.12.3 Compiled Qt version: 5.12.4 Runtime Qt version: 5.12.8
Attachments (1)
{kind=link}
{kind=link}
Change History (13)
comment:1 by , 6 years ago
| Cc: | added |
|---|---|
| Component: | Unassigned → Structure Comparison |
| Owner: | set to |
| Platform: | → all |
| Project: | → ChimeraX |
| Status: | new → assigned |
| Summary: | ChimeraX bug report submission → Maximum common substructure |
| Type: | defect → enhancement |
follow-up: 2 comment:2 by , 6 years ago
I'm just working on a port of the code from https://github.com/jamestrimble/mcsplit-induced-subgraph-isomorphism (https://www.ijcai.org/Proceedings/2017/99). It's fairly simple in that nodes can only be labelled with ints - but that's good enough for my purposes (just use the atomic number). Just got it to compile with some simple Python bindings... now to see if it will do the job. The RDKit implementation is of course much more tailored towards chemistry (for instance, it can take bond orders into account, and allow you to decide whether partial rings are allowed). Still, this will probably do for the time being (assuming it works, of course). On 2020-06-17 16:49, ChimeraX wrote:
comment:4 by , 6 years ago
When you tried to match phosphatidylcholine to phosphatidylethanolamine with networkx did the graph nodes have the element number which must be matched? I can't imagine how that match could spend a lot of time unless all the graph nodes were considered equivalent.
follow-up: 5 comment:5 by , 6 years ago
Element name, actually. It *does* get the right answer on smaller cases - it's just *really* getting bogged down somewhere. On 2020-06-17 17:53, ChimeraX wrote:
follow-up: 6 comment:6 by , 6 years ago
Hmm... the McSplit implementation is running without crashing, but at
the moment is only giving an answer for a subset of cases (for the rest
it returns an empty set).
The NetworkX ISMAGS implementation is slow (at least, when hydrogens are
present) because it's doing an absolutely insane amount of sorting... if
I have:
{{{
class AtomProxy:
def __init__(self, atom):
self.atom = atom
def __lt__(self, other):
return self.atom.name < other.atom.name
def residue_graph(residue):
'''
Make a :class:`networkx.Graph` representing the connectivity of a
residue's
atoms.
'''
import networkx as nx
rn = nx.Graph()
atoms = residue.atoms
proxies = [AtomProxy(atom) for atom in atoms]
proxy_map = {atom: proxy for atom, proxy in zip(atoms, proxies)}
rn.add_nodes_from(proxies)
nx.set_node_attributes(rn, {a: {'element': e, 'name': n} for a, e, n
in zip(proxies, atoms.element_names, atoms.names)})
rn.add_edges_from([[proxy_map[a] for a in b.atoms] for b in
atoms.intra_bonds])
return rn
def find_maximal_isomorphous_fragment(residue_graph, template_graph,
match_by='name'):
'''
When a residue doesn't quite match its template, there can be
various
explanations:
- the residue is incomplete (e.g. lacking hydrogens, or actually
truncated)
- the hydrogen addition has misfired, adding too many or too few
hydrogens
- this isn't actually the correct template for the residue
This method compares the graph representations of residue and
template to
find the maximal overlap, returning a dict mapping nodes in the
larger
to their equivalent in the smaller.
Args:
* residue_graph
- a :class:`networkx.Graph` object representing the residue
atoms
present in the model (e.g. as built by
:func:`residue_graph`)
* template_graph
- a :class:`networkx.Graph` object representing the residue
atoms
present in the template (e.g. as built by
:func:`template_graph`)
* match_by (default: 'name')
- atomic property to match nodes by (one of 'element' or
'name')
Returns:
* matched_atoms (dict)
- mapping of residue atoms to matching template atoms
* residue_extra_atoms (list)
- atoms that are in the residue but were not matched to the
template
(if the residue is disconnected into two or more
fragments, all
atoms that aren't in the largest will be found here).
* template_extra_atoms (list)
- atoms in the template that aren't in the residue.
'''
import networkx as nx
from networkx import isomorphism as iso
max_nodes = 0
largest_sg = None
for cg in nx.connected.connected_components(residue_graph):
if len(cg) > max_nodes:
max_nodes = len(cg)
largest_sg = residue_graph.subgraph(cg)
if len(largest_sg) > len(template_graph):
residue_larger = True
else:
residue_larger = False
# gm = iso.GraphMatcher(lg, largest_sg,
# node_match=iso.categorical_node_match(match_by, None))
if residue_larger:
ismags = iso.ISMAGS(largest_sg, template_graph,
node_match=iso.categorical_node_match(match_by, ''))
else:
ismags = iso.ISMAGS(template_graph, largest_sg,
node_match=iso.categorical_node_match(match_by, ''))
subgraphs = ismags.largest_common_subgraph(symmetry=True)
# There may be multiple symmetry-equivalent graphs when we match by
element,
# so just take the first one
for sg in subgraphs:
break
# Convert from the AtomProxy objects back to the underlying atoms
if residue_larger:
sg = {key.atom: val.atom for key, val in sg.items()}
else:
sg = {val.atom: key.atom for key, val in sg.items()}
residue_atoms = set(a.atom for a in largest_sg.nodes)
template_atoms = set(a.atom for a in template_graph.nodes)
residue_extra_atoms = residue_atoms.difference(set(sg.keys()))
template_extra_atoms = template_atoms.difference(set(sg.values()))
return sg, list(residue_extra_atoms), list(template_extra_atoms)
# pe is 3PE (phosphatidylethanolamine), pc is PC1 (phosphatidylcholine)
rpe = residue_graph(pe)
rpc = residue_graph(pc)
%prun find_maximal_isomorphous_fragment(rpe, prc, 'name')
269450443 function calls (268236996 primitive calls) in 180.962
seconds
Ordered by: internal time
ncalls tottime percall cumtime percall filename:lineno(function)
263984135 107.965 0.000 107.965 0.000
template_utils.py:79(__lt__)
4 55.440 13.860 163.405 40.851 {built-in method
builtins.sorted}
1167607 8.983 0.000 8.983 0.000
ismags.py:925(_remove_node)
1167873 3.757 0.000 3.757 0.000 {method 'add' of 'set'
objects}
}}}
... that's a quarter *billion* calls to AtomProxy.__lt__. Geezus.
On 2020-06-17 17:13, ChimeraX wrote:
follow-up: 7 comment:7 by , 6 years ago
Well... there's my problem. I wrapped the wrong damn library. He's got multiple different implementations, and this one's designed to solve a slightly different problem than the one in the paper (as the name says, it solves the induced subgraph isomorphism problem - https://en.wikipedia.org/wiki/Induced_subgraph_isomorphism_problem - rather than the maximum common induced subgraph - https://en.wikipedia.org/wiki/Maximum_common_induced_subgraph). Sigh... the good news is that it was very straightforward to adapt. I've emailed him to ask directly which is the best of his many repositories to use. On 2020-06-17 19:24, ChimeraX wrote:
follow-up: 8 comment:8 by , 6 years ago
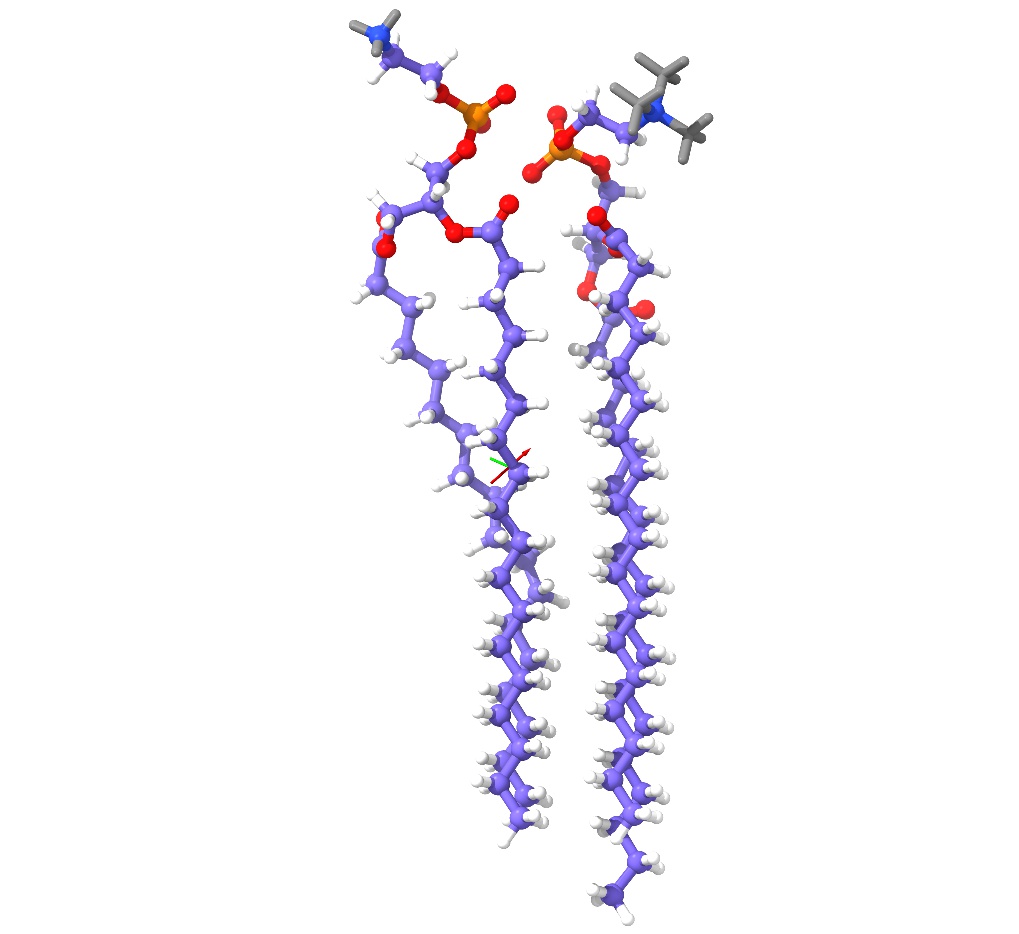
James very helpfully pointed me to the correct code, and it's now working mostly quite nicely (attached result for phosphatidylserine vs. phosphatidylcholine - grey atoms are those identified as not matching). One niggling issue that I'm asking him about: for this particular case its logging showed that it found the correct answer almost immediately (in a handful of milliseconds), but it didn't actually return until forced by its timeout (and hung indefinitely if the timeout parameter was set to zero). Even so, it beats NetworkX hands down - for the same scenario (atoms labelled by element rather than by name) it took about 20 minutes! On 2020-06-17 20:57, ChimeraX wrote:
comment:10 by , 6 years ago
Working implementation at
https://github.com/tristanic/isolde/tree/master/isolde/src/graph. A
basic test script looks like:
{{{
from time import time
start_time = time()
from chimerax.atomic import selected_residues
r1, r2 = selected_residues(session)
from chimerax.isolde.graph import test_make_graph
r1g = test_make_graph(r1)
r2g = test_make_graph(r2)
session.selection.clear()
i1, i2, timed_out = r1g.maximum_common_subgraph(r2g, timeout=0.25)
r1.atoms[i1].selected=True
r2.atoms[i2].selected=True
if timed_out:
print('McSplit timed out')
end_time = time()
print('Finding maximum common induced subgraph took {:.3f}
seconds'.format(end_time-start_time))
}}}
James added some extra code to improve the performance on the lipid
case, and that now takes ~25 ms. It still gets bogged down on multi-ring
systems (e.g. matching heme (HEM) to bacteriochlorophyll (BCL)) -
similar to the lipids, it finds a good (actually, what looks like the
optimal) solution very quickly, but then spends a very long time trying
to prove it. That's probably not a big deal - as long as the timeout is
set to something reasonable, it'll still return the best solution it's
found.
I'm open on suggestions as to where/how to put this. It seems like a
general-purpose utility that would find lots of applications outside of
ISOLDE (e.g. aligning ligands based on their common structure) so it
would probably make most sense to split it into a separate bundle?
He's also said he'd be happy to write a pure-Python implementation of
the same algorithm. I suggested if he was going to do that it might be
best to contribute it to NetworkX, since their existing MCS
implementation really does seem pretty terrible. We'll see what comes of
that.
On 2020-06-18 16:55, ChimeraX wrote:
follow-up: 10 comment:11 by , 6 years ago
Update: this is working quite neatly for most cases. I can reliably interconvert between phosphatidylcholine and phosphatidylethanolamine in about 40ms (matching, renaming all atoms, deleting superfluous ones and adding the new ones), or cycle between ADP, ATP, GDP, GTP etc.. Still a few remaining issues: - matching HEM to BCL fails pretty badly when hydrogens are included, but is really fast without them. I've done some quick experiments with "rolling" hydrogens into their neighbors, labelling the heavy atom node as (element_number*10 + num_hydrogens) - seems to make things *much* more robust. I'll try to make this happen on the C++ side so the Python code doesn't have to worry about it... - atoms are added using internal geometry calculated from the template, but no attempt is currently made to avoid clashes. So turning ADP into ATP, for example, can cause a pretty severe clash depending on which of the three equivalent terminal oxygens gets the new phosphate. Dealing with that will take some extra work (open to suggestions there!). On 2020-06-18 17:23, ChimeraX wrote:
follow-up: 11 comment:12 by , 3 years ago
| Resolution: | → worksforme |
|---|---|
| Status: | assigned → closed |
I still need to revisit this at some time in the future to make it stereospecific, but what's there works well enough for now.
Note:
See TracTickets
for help on using tickets.
I have not needed maximum common substructure algorithms so far, so have nothing to contribute that wouldn't be regurgitated Google-fu.