#18396 closed defect (not a bug)
Ribbon does not include non-standard residues
| Reported by: | Owned by: | Tom Goddard | |
|---|---|---|---|
| Priority: | normal | Milestone: | |
| Component: | Depiction | Version: | |
| Keywords: | Cc: | Greg Couch, Eric Pettersen | |
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
The following bug report has been submitted:
Platform: Windows-10-10.0.26100
ChimeraX Version: 1.10 (2025-06-26 08:57:52 UTC)
Description
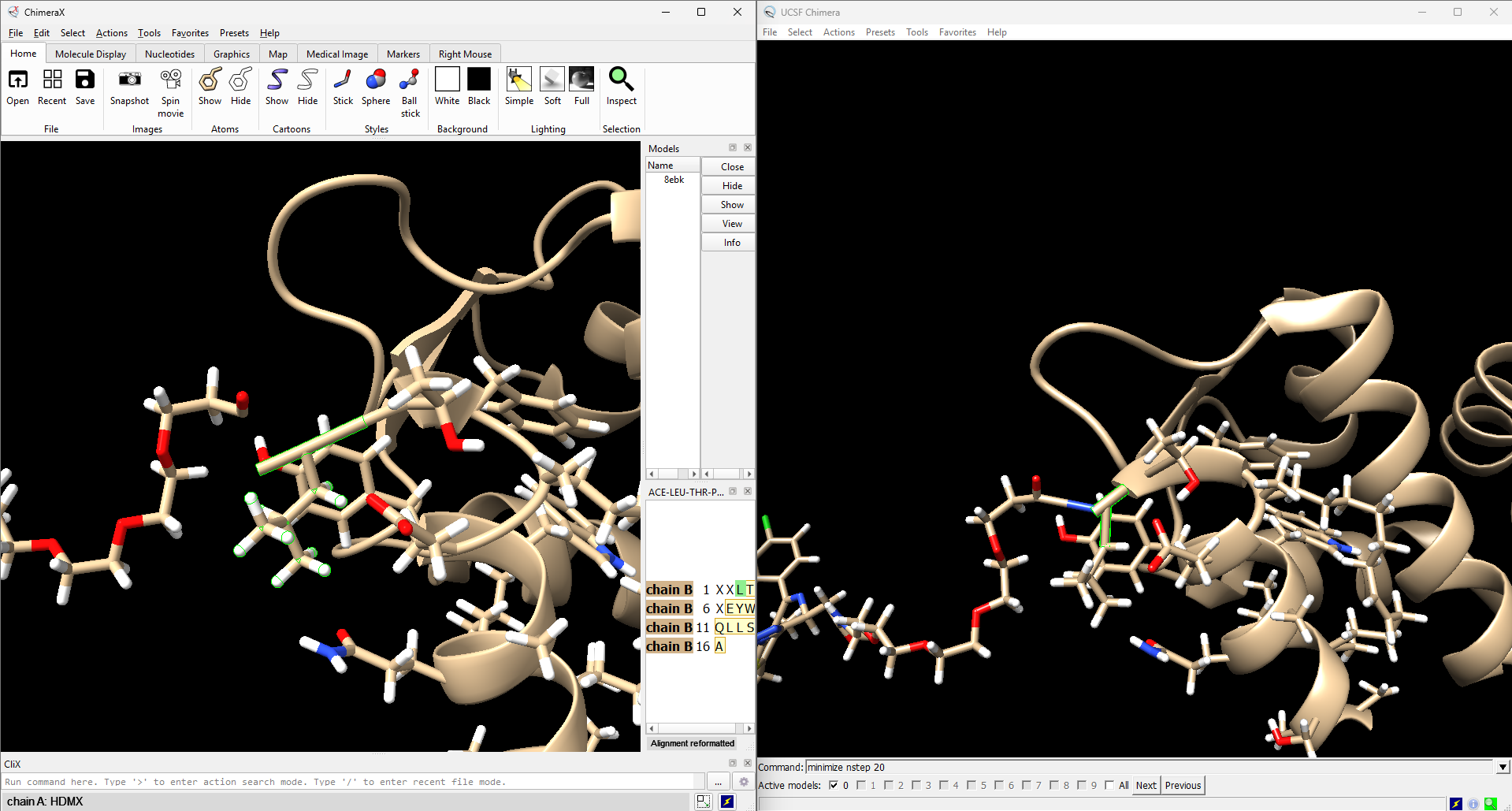
Hello, I was working on PROTAC structure complex (PDB ID: 8EBK), this is where i see that the leucine (residue id 3) doesnot appear to bind with residue id 2, 3V3 in ChimeraX. when I compared the same in Chimera program, there is no issue and the bond is also visible linking to residues. portion is highlighted in the attached image for both ChimeraX and Chimera. i would highly appreciate the resolution of this matter
Log:
> ui tool hide "command line interface"
> ui tool show clix
UCSF ChimeraX version: 1.10 (2025-06-26)
© 2016-2025 Regents of the University of California. All rights reserved.
How to cite UCSF ChimeraX
> open 8ebk format mmcif fromDatabase pdb
8ebk title:
Crystal Structure Analysis of xHDMX in complex with the stapled peptide PROTAC
analog [more info...]
Chain information for 8ebk #1
---
Chain | Description
A | HDMX
B | ACE-LEU-THR-PHE-0EH-GLU-TYR-TRP-ALA-GLN-LEU-MK8-SER-ALA-ALA
Non-standard residues in 8ebk #1
---
0EH — (2R)-2-amino-2-methylnonanoic acid
3V3 — 1-amino-3,6,9,12-tetraoxapentadecan-15-oic acid
WG0 —
[(6S,10P)-4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl]acetic
acid
29 atoms have alternate locations. Control/examine alternate locations with
Altloc Explorer [start tool...] or the altlocs command.
> select /B
196 atoms, 172 bonds, 48 residues, 1 model selected
> show sel atoms
> sequence chain #1/B
Alignment identifier is 1/B
> select /B:3
8 atoms, 7 bonds, 1 residue, 1 model selected
> select /B:3
8 atoms, 7 bonds, 1 residue, 1 model selected
> select ::name="HOH"
119 atoms, 119 residues, 1 model selected
> delete atoms sel
> delete bonds sel
> dockprep
Starting dock prep
Deleting solvent
Deleting non-metal-complex ions
Deleting non-current alt locs
Summary of feedback from adding hydrogens to 8ebk #1
---
notes | Termini for 8ebk (#1) chain A determined from SEQRES records
Termini for 8ebk (#1) chain B determined from SEQRES records
Chain-initial residues that are actual N termini: /B WG0 1
Chain-initial residues that are not actual N termini: /A GLY 21
Chain-final residues that are actual C termini: /B ALA 16
Chain-final residues that are not actual C termini: /A LEU 106
88 hydrogen bonds
Adding 'H' to /A GLY 21
/A LEU 106 is not terminus, removing H atom from 'C'
870 hydrogens added
Using Amber 20 recommended default charges and atom types for standard
residues
Assigning partial charges to residue WG0+3V3 (net charge +0) with am1-bcc
method
Running ANTECHAMBER command: C:/Program Files/ChimeraX
1.10/bin/amber20/bin/antechamber -ek qm_theory='AM1', -i
C:\Users\TUSHA_~1\AppData\Local\Temp\tmpoen7gylx\ante.in.mol2 -fi mol2 -o
C:\Users\TUSHA_~1\AppData\Local\Temp\tmpoen7gylx\ante.out.mol2 -fo mol2 -c bcc
-nc 0 -j 5 -s 2 -dr n
(WG0+3V3) ``
(WG0+3V3) `Welcome to antechamber 20.0: molecular input file processor.`
(WG0+3V3) ``
(WG0+3V3) `Info: Finished reading file
(C:\Users\TUSHA_~1\AppData\Local\Temp\tmpoen7gylx\ante.in.mol2); atoms read
(86), bonds read (89).`
(WG0+3V3) `Info: Determining atomic numbers from atomic symbols which are case
sensitive.`
(WG0+3V3) `Running: "C:/Program Files/ChimeraX 1.10/bin/amber20/bin/bondtype"
-j part -i ANTECHAMBER_BOND_TYPE.AC0 -o ANTECHAMBER_BOND_TYPE.AC -f ac`
(WG0+3V3) `bash.exe: warning: could not find /tmp, please create!`
(WG0+3V3) ``
(WG0+3V3) ``
(WG0+3V3) `Running: "C:/Program Files/ChimeraX 1.10/bin/amber20/bin/atomtype"
-i ANTECHAMBER_AC.AC0 -o ANTECHAMBER_AC.AC -p gaff`
(WG0+3V3) `bash.exe: warning: could not find /tmp, please create!`
(WG0+3V3) `Info: Total number of electrons: 350; net charge: 0`
(WG0+3V3) ``
(WG0+3V3) `Running: "C:/Program Files/ChimeraX 1.10/bin/amber20/bin/sqm" -O -i
sqm.in -o sqm.out`
(WG0+3V3) `bash.exe: warning: could not find /tmp, please create!`
(WG0+3V3) ``
(WG0+3V3) `Running: "C:/Program Files/ChimeraX 1.10/bin/amber20/bin/am1bcc" -i
ANTECHAMBER_AM1BCC_PRE.AC -o ANTECHAMBER_AM1BCC.AC -f ac -p "C:/Program
Files/ChimeraX 1.10/bin/amber20/dat/antechamber/BCCPARM.DAT" -s 2 -j 1`
(WG0+3V3) `bash.exe: warning: could not find /tmp, please create!`
(WG0+3V3) ``
(WG0+3V3) `Running: "C:/Program Files/ChimeraX 1.10/bin/amber20/bin/atomtype"
-f ac -p bcc -o ANTECHAMBER_AM1BCC.AC -i ANTECHAMBER_AM1BCC_PRE.AC`
(WG0+3V3) `bash.exe: warning: could not find /tmp, please create!`
(WG0+3V3) ``
Charges for residue WG0+3V3 determined
Assigning partial charges to residue 0EH+MK8 (net charge +0) with am1-bcc
method
Running ANTECHAMBER command: C:/Program Files/ChimeraX
1.10/bin/amber20/bin/antechamber -ek qm_theory='AM1', -i
C:\Users\TUSHA_~1\AppData\Local\Temp\tmpmmt4n9h3\ante.in.mol2 -fi mol2 -o
C:\Users\TUSHA_~1\AppData\Local\Temp\tmpmmt4n9h3\ante.out.mol2 -fo mol2 -c bcc
-nc 0 -j 5 -s 2 -dr n
(0EH+MK8) ``
(0EH+MK8) `Welcome to antechamber 20.0: molecular input file processor.`
(0EH+MK8) ``
(0EH+MK8) `Info: Finished reading file
(C:\Users\TUSHA_~1\AppData\Local\Temp\tmpmmt4n9h3\ante.in.mol2); atoms read
(73), bonds read (72).`
(0EH+MK8) `Info: Determining atomic numbers from atomic symbols which are case
sensitive.`
(0EH+MK8) `Running: "C:/Program Files/ChimeraX 1.10/bin/amber20/bin/bondtype"
-j part -i ANTECHAMBER_BOND_TYPE.AC0 -o ANTECHAMBER_BOND_TYPE.AC -f ac`
(0EH+MK8) `bash.exe: warning: could not find /tmp, please create!`
(0EH+MK8) ``
(0EH+MK8) ``
(0EH+MK8) `Running: "C:/Program Files/ChimeraX 1.10/bin/amber20/bin/atomtype"
-i ANTECHAMBER_AC.AC0 -o ANTECHAMBER_AC.AC -p gaff`
(0EH+MK8) `bash.exe: warning: could not find /tmp, please create!`
(0EH+MK8) `Info: Total number of electrons: 240; net charge: 0`
(0EH+MK8) ``
(0EH+MK8) `Running: "C:/Program Files/ChimeraX 1.10/bin/amber20/bin/sqm" -O -i
sqm.in -o sqm.out`
(0EH+MK8) `bash.exe: warning: could not find /tmp, please create!`
(0EH+MK8) ``
(0EH+MK8) `Running: "C:/Program Files/ChimeraX 1.10/bin/amber20/bin/am1bcc" -i
ANTECHAMBER_AM1BCC_PRE.AC -o ANTECHAMBER_AM1BCC.AC -f ac -p "C:/Program
Files/ChimeraX 1.10/bin/amber20/dat/antechamber/BCCPARM.DAT" -s 2 -j 1`
(0EH+MK8) `bash.exe: warning: could not find /tmp, please create!`
(0EH+MK8) ``
(0EH+MK8) `Running: "C:/Program Files/ChimeraX 1.10/bin/amber20/bin/atomtype"
-f ac -p bcc -o ANTECHAMBER_AM1BCC.AC -i ANTECHAMBER_AM1BCC_PRE.AC`
(0EH+MK8) `bash.exe: warning: could not find /tmp, please create!`
(0EH+MK8) ``
Charges for residue 0EH+MK8 determined
1 structure has non-integral total charge.
Details in log.
Here are the structures will non-integral total charge along with the
particular residues from those structures with non-integral total charge:
8ebk #1: /B WG0 1: 0.0119995, /B 3V3 2: -0.0189995, /B 0EH 6: 0.00934653, and
/B MK8 13: -0.0123465
Dock prep finished
Drag select of 1 residues
> help help:contact.html
> select /B
320 atoms, 328 bonds, 16 residues, 1 model selected
> select /B:3
19 atoms, 18 bonds, 1 residue, 1 model selected
> select /B:3
19 atoms, 18 bonds, 1 residue, 1 model selected
> select /B:3
19 atoms, 18 bonds, 1 residue, 1 model selected
> select /B:3
19 atoms, 18 bonds, 1 residue, 1 model selected
OpenGL version: 3.3.0 NVIDIA 572.83
OpenGL renderer: NVIDIA GeForce RTX 4090/PCIe/SSE2
OpenGL vendor: NVIDIA Corporation
Python: 3.11.4
Locale: en_IN.cp1252
Qt version: PyQt6 6.8.1, Qt 6.8.2
Qt runtime version: 6.8.2
Qt platform: windows
Manufacturer: HP
Model: OMEN by HP 45L Gaming Desktop GT22-1xxx
OS: Microsoft Windows 11 Home Single Language (Build 26100)
Memory: 68,388,151,296
MaxProcessMemory: 137,438,953,344
CPU: 32 13th Gen Intel(R) Core(TM) i9-13900K
OSLanguage: en-US
Installed Packages:
alabaster: 1.0.0
appdirs: 1.4.4
asttokens: 3.0.0
auditwheel: 6.4.0
babel: 2.17.0
beautifulsoup4: 4.13.3
blockdiag: 3.0.0
blosc2: 3.5.0
build: 1.2.2.post1
certifi: 2025.6.15
cftime: 1.6.4.post1
charset-normalizer: 3.4.2
ChimeraX-AddCharge: 1.5.19
ChimeraX-AddH: 2.2.7
ChimeraX-AlignmentAlgorithms: 2.0.2
ChimeraX-AlignmentHdrs: 3.6.1
ChimeraX-AlignmentMatrices: 2.1
ChimeraX-Alignments: 2.20.2
ChimeraX-AlphaFold: 1.0.1
ChimeraX-AltlocExplorer: 1.1.2
ChimeraX-AmberInfo: 1.0
ChimeraX-Aniso: 1.1.4
ChimeraX-Arrays: 1.1
ChimeraX-Atomic: 1.60.7
ChimeraX-AtomicLibrary: 14.1.18
ChimeraX-AtomSearch: 2.0.1
ChimeraX-AxesPlanes: 2.4
ChimeraX-BasicActions: 1.1.3
ChimeraX-BILD: 1.0
ChimeraX-BlastProtein: 3.0.0
ChimeraX-Boltz: 1.0
ChimeraX-BondRot: 2.0.4
ChimeraX-BugReporter: 1.0.2
ChimeraX-BuildStructure: 2.13.1
ChimeraX-Bumps: 1.0
ChimeraX-BundleBuilder: 1.5.1
ChimeraX-ButtonPanel: 1.0.1
ChimeraX-CageBuilder: 1.0.1
ChimeraX-CellPack: 1.0
ChimeraX-Centroids: 1.4
ChimeraX-ChangeChains: 1.1
ChimeraX-CheckWaters: 1.5
ChimeraX-ChemGroup: 2.0.2
ChimeraX-Clashes: 2.3
ChimeraX-clix: 0.2.3
ChimeraX-ColorActions: 1.0.5
ChimeraX-ColorGlobe: 1.0
ChimeraX-ColorKey: 1.5.8
ChimeraX-CommandLine: 1.3
ChimeraX-ConnectStructure: 2.0.1
ChimeraX-Contacts: 1.0.1
ChimeraX-Core: 1.10
ChimeraX-CoreFormats: 1.2
ChimeraX-coulombic: 1.4.5
ChimeraX-Crosslinks: 1.0
ChimeraX-Crystal: 1.0
ChimeraX-CrystalContacts: 1.0.1
ChimeraX-DataFormats: 1.2.4
ChimeraX-Dicom: 1.2.7
ChimeraX-DistMonitor: 1.4.2
ChimeraX-DockPrep: 1.1.4
ChimeraX-Dssp: 2.0
ChimeraX-EMDB-SFF: 1.0
ChimeraX-ESMFold: 1.0
ChimeraX-FileHistory: 1.0.1
ChimeraX-FunctionKey: 1.0.1
ChimeraX-Geometry: 1.3
ChimeraX-gltf: 1.0
ChimeraX-Graphics: 1.4.1
ChimeraX-Hbonds: 2.5.1
ChimeraX-Help: 1.3
ChimeraX-HKCage: 1.3
ChimeraX-IHM: 1.1
ChimeraX-ImageFormats: 1.2
ChimeraX-IMOD: 1.0
ChimeraX-IO: 1.0.3
ChimeraX-ItemsInspection: 1.0.1
ChimeraX-IUPAC: 1.0
ChimeraX-KVFinder: 1.6.2
ChimeraX-Label: 1.1.14
ChimeraX-ListInfo: 1.2.2
ChimeraX-Log: 1.2
ChimeraX-LookingGlass: 1.1
ChimeraX-Maestro: 1.9.1
ChimeraX-Map: 1.3
ChimeraX-MapData: 2.0
ChimeraX-MapEraser: 1.0.1
ChimeraX-MapFilter: 2.0.1
ChimeraX-MapFit: 2.0
ChimeraX-MapSeries: 2.1.1
ChimeraX-Markers: 1.0.1
ChimeraX-Mask: 1.0.2
ChimeraX-MatchMaker: 2.2.2
ChimeraX-MCopy: 1.0
ChimeraX-MDcrds: 2.10.1
ChimeraX-MedicalToolbar: 1.1
ChimeraX-Meeting: 1.0.1
ChimeraX-MLP: 1.1.1
ChimeraX-mmCIF: 2.16
ChimeraX-MMTF: 2.2
ChimeraX-ModelArchive: 1.0
ChimeraX-Modeller: 1.5.19
ChimeraX-ModelPanel: 1.5.1
ChimeraX-ModelSeries: 1.0.1
ChimeraX-Mol2: 2.0.3
ChimeraX-Mole: 1.0
ChimeraX-Morph: 1.0.2
ChimeraX-MouseModes: 1.2
ChimeraX-Movie: 1.0
ChimeraX-MutationScores: 1.0
ChimeraX-Neuron: 1.0
ChimeraX-Nifti: 1.2
ChimeraX-NMRSTAR: 1.0.2
ChimeraX-NRRD: 1.2
ChimeraX-Nucleotides: 2.0.3
ChimeraX-OpenCommand: 1.14.1
ChimeraX-OrthoPick: 1.0.1
ChimeraX-PDB: 2.7.10
ChimeraX-PDBBio: 1.0.1
ChimeraX-PDBLibrary: 1.0.4
ChimeraX-PDBMatrices: 1.0
ChimeraX-PickBlobs: 1.0.1
ChimeraX-Positions: 1.0
ChimeraX-PresetMgr: 1.1.3
ChimeraX-ProfileGrids: 1.1.2
ChimeraX-PubChem: 2.2
ChimeraX-ReadPbonds: 1.0.1
ChimeraX-Registration: 1.1.2
ChimeraX-RemoteControl: 1.0
ChimeraX-RenderByAttr: 1.6.3
ChimeraX-RenumberResidues: 1.1
ChimeraX-ResidueFit: 1.0.1
ChimeraX-RestServer: 1.3.1
ChimeraX-RNALayout: 1.0
ChimeraX-RotamerLibMgr: 4.0
ChimeraX-RotamerLibsDunbrack: 2.0
ChimeraX-RotamerLibsDynameomics: 2.0
ChimeraX-RotamerLibsRichardson: 2.0
ChimeraX-SaveCommand: 1.5.1
ChimeraX-SchemeMgr: 1.0
ChimeraX-SDF: 2.0.3
ChimeraX-Segger: 1.0
ChimeraX-Segment: 1.0.1
ChimeraX-Segmentations: 3.5.7
ChimeraX-SelInspector: 1.0
ChimeraX-SeqView: 2.17.1
ChimeraX-Shape: 1.1
ChimeraX-Shell: 1.0.1
ChimeraX-Shortcuts: 1.2.1
ChimeraX-ShowSequences: 1.0.3
ChimeraX-SideView: 1.0.1
ChimeraX-SimilarStructures: 1.0.1
ChimeraX-Smiles: 2.1.2
ChimeraX-SmoothLines: 1.0
ChimeraX-SpaceNavigator: 1.0
ChimeraX-StdCommands: 1.19.1
ChimeraX-STL: 1.0.1
ChimeraX-Storm: 1.0
ChimeraX-StructMeasure: 1.2.1
ChimeraX-Struts: 1.0.1
ChimeraX-Surface: 1.0.1
ChimeraX-SwapAA: 2.0.1
ChimeraX-SwapRes: 2.5.2
ChimeraX-TapeMeasure: 1.0
ChimeraX-TaskManager: 1.0
ChimeraX-Test: 1.0
ChimeraX-Toolbar: 1.2.3
ChimeraX-ToolshedUtils: 1.2.4
ChimeraX-Topography: 1.0
ChimeraX-ToQuest: 1.0
ChimeraX-Tug: 1.0.1
ChimeraX-UI: 1.45.2
ChimeraX-Umap: 1.0
ChimeraX-uniprot: 2.3.1
ChimeraX-UnitCell: 1.0.1
ChimeraX-ViewDockX: 1.4.4
ChimeraX-VIPERdb: 1.0
ChimeraX-Vive: 1.1
ChimeraX-VolumeMenu: 1.0.1
ChimeraX-vrml: 1.0
ChimeraX-VTK: 1.0
ChimeraX-WavefrontOBJ: 1.0
ChimeraX-WebCam: 1.0.2
ChimeraX-WebServices: 1.1.5
ChimeraX-Zone: 1.0.1
colorama: 0.4.6
comm: 0.2.2
comtypes: 1.4.10
contourpy: 1.3.2
coverage: 7.9.1
cxservices: 1.2.3
cycler: 0.12.1
Cython: 3.0.12
debugpy: 1.8.14
decorator: 5.2.1
docutils: 0.21.2
executing: 2.2.0
filelock: 3.18.0
fonttools: 4.58.4
funcparserlib: 2.0.0a0
glfw: 2.9.0
grako: 3.16.5
h5py: 3.14.0
html2text: 2024.2.26
idna: 3.10
ihm: 2.2
imagecodecs: 2024.6.1
imagesize: 1.4.1
iniconfig: 2.1.0
ipykernel: 6.29.5
ipython: 8.26.0
ipywidgets: 8.1.7
jedi: 0.19.1
Jinja2: 3.1.6
jupyter_client: 8.6.3
jupyter_core: 5.8.1
jupyterlab_widgets: 3.0.15
kiwisolver: 1.4.8
line_profiler: 4.2.0
lxml: 5.3.1
lz4: 4.4.4
MarkupSafe: 3.0.2
matplotlib: 3.10.1
matplotlib-inline: 0.1.7
msgpack: 1.1.0
ndindex: 1.10.0
nest-asyncio: 1.6.0
netCDF4: 1.6.5
networkx: 3.3
nibabel: 5.2.0
nptyping: 2.5.0
numexpr: 2.11.0
numpy: 1.26.4
nvidia-cuda-cupti-cu12: 12.9.79
nvidia-cuda-nvcc-cu12: 12.9.86
nvidia-cuda-nvrtc-cu12: 12.9.86
nvidia-cuda-runtime-cu12: 12.9.79
nvidia-cufft-cu12: 11.4.1.4
nvidia-nvjitlink-cu12: 12.9.86
OpenMM: 8.2.0
OpenMM-CUDA-12: 8.2.0
openvr: 1.26.701
packaging: 24.2
ParmEd: 4.2.2
parso: 0.8.4
pep517: 0.13.1
pickleshare: 0.7.5
pillow: 10.4.0
pip: 25.0.1
pkginfo: 1.11.1
platformdirs: 4.3.8
pluggy: 1.6.0
prompt_toolkit: 3.0.51
psutil: 7.0.0
pure_eval: 0.2.3
py-cpuinfo: 9.0.0
pycollada: 0.8
pydicom: 2.4.4
pyelftools: 0.32
Pygments: 2.18.0
pynmrstar: 3.3.5
pynrrd: 1.0.0
PyOpenGL: 3.1.9
PyOpenGL-accelerate: 3.1.9
pyopenxr: 1.1.4501
pyparsing: 3.2.3
pyproject_hooks: 1.2.0
PyQt6-commercial: 6.8.1
PyQt6-Qt6: 6.8.2
PyQt6-WebEngine-commercial: 6.8.0
PyQt6-WebEngine-Qt6: 6.8.2
PyQt6_sip: 13.10.0
pytest: 8.4.1
pytest-cov: 6.2.1
python-dateutil: 2.9.0.post0
pytz: 2025.2
pywin32: 310
pyzmq: 27.0.0
qtconsole: 5.5.2
QtPy: 2.4.3
qtshim: 1.1
RandomWords: 0.4.0
requests: 2.32.3
roman-numerals-py: 3.1.0
scipy: 1.14.0
setuptools: 78.1.0
sfftk-rw: 0.8.1
six: 1.16.0
snowballstemmer: 3.0.1
sortedcontainers: 2.4.0
soupsieve: 2.7
Sphinx: 8.2.3
sphinx-autodoc-typehints: 3.1.0
sphinxcontrib-applehelp: 2.0.0
sphinxcontrib-blockdiag: 3.0.0
sphinxcontrib-devhelp: 2.0.0
sphinxcontrib-htmlhelp: 2.1.0
sphinxcontrib-jsmath: 1.0.1
sphinxcontrib-qthelp: 2.0.0
sphinxcontrib-serializinghtml: 2.0.0
stack-data: 0.6.3
superqt: 0.7.1
tables: 3.10.2
tcia_utils: 1.5.1
tifffile: 2025.3.13
tinyarray: 1.2.4
tornado: 6.5.1
traitlets: 5.14.3
typing_extensions: 4.14.0
tzdata: 2025.2
urllib3: 2.5.0
wcwidth: 0.2.13
webcolors: 24.11.1
wheel: 0.45.1
wheel-filename: 1.4.2
widgetsnbextension: 4.0.14
WMI: 1.5.1
File attachment: Screenshot 2025-08-08 121834.png
{kind=link}
{kind=link}
Attachments (1)
Change History (3)
by , 12 months ago
| Attachment: | Screenshot 2025-08-08 121834.png added |
|---|
comment:1 by , 12 months ago
| Cc: | added |
|---|---|
| Component: | Unassigned → Depiction |
| Owner: | set to |
| Platform: | → all |
| Project: | → ChimeraX |
| Status: | new → assigned |
| Summary: | ChimeraX bug report submission → Ribbon does not include non-standard residues |
Reported by Tushar Gupta
comment:2 by , 12 months ago
| Resolution: | → not a bug |
|---|---|
| Status: | assigned → closed |
Residue 2 in chain B of 8EBK has no C-alpha atom (name CA) and ribbons compute a spline through C-alpha atoms. So residues that don't have a CA atom are not connected by ribbons. I am quite amazed that Chimera can connect it!
You can rename the carbon atom of residue 2 that is in the C-alpha position from its current name C14 to CA and then the ribbon will connect it. Here is the ChimeraX command to rename that atom
setattr /B:2@C14 a name CA
You will need to hide and reshow ribbons to recompute the ribbon after that change. There is also an atom named "N" at the end of the residue 2 side chain that confuses the ribbon code because it thinks that is the backbone nitrogen, so it draws an absurd blue "tether" to it. To get rid of that you could rename that nitrogen
setattr /B:2@N a name N1
Again hide and reshow ribbons to fix the ribbon appearance.
I'm not going to try to make the ChimeraX code handle this case automatically. I'm surprised Chimera can do it. I think protein ribbons should have a ribbon backbone including C, CA, and N atoms, or in minimal CA-only structures just CA. You can't just put any side group at the end having a carbon atom and expect the ribbon to extend to it.
Added by email2trac