#17918 closed defect (not a bug)
Difference in secondary structure display between Mol* 3D and ChimeraX
| Reported by: | Owned by: | Eric Pettersen | |
|---|---|---|---|
| Priority: | normal | Milestone: | |
| Component: | Structure Analysis | Version: | |
| Keywords: | Cc: | ||
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
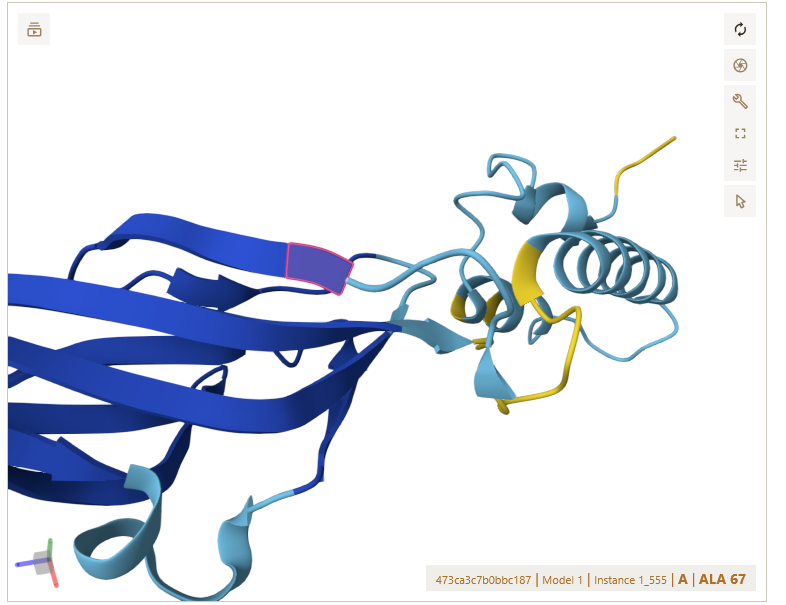
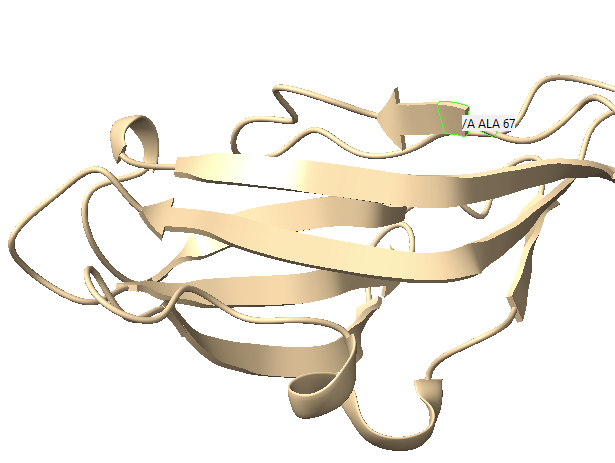
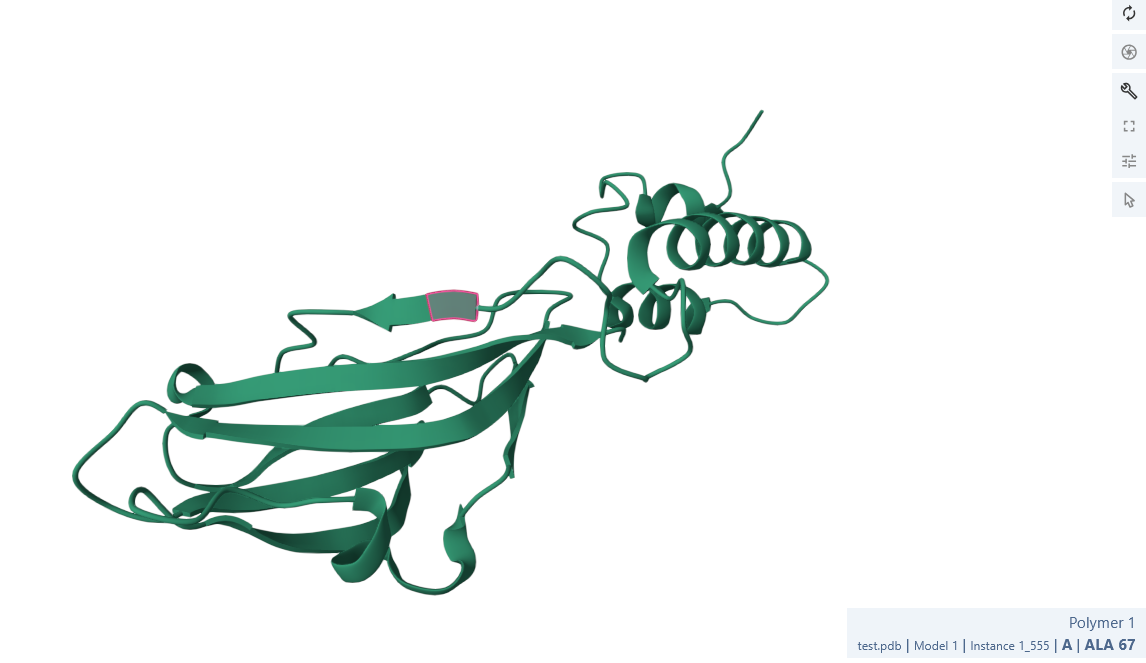
Hello, upon importing an Alphafold3 prediction (which as far as I can tell used the PDB Mol*3D viewer to display results) into ChimeraX 1.1 and trying to draw a fold diagram based on it, I noticed that ChimeraX was missing and/or shortening the display of certain secondary structures. I have attached screenshots for Mol*3d and ChimeraX: [cid:fd040d24-4c98-469e-8881-7da7c3a649b8] [cid:5b8b968d-769e-4b61-96a3-c6c16f22a8f9] As you can see the beta sheet starting at ALA67 (highlighted in both pictures) is much shorter in ChimeraX, and the beta sheet after the turn is missing altogether. Saving the file from ChimeraX and re-uploading it to the PDB viewer gives the chimeraX result: [cid:46d89741-fcf9-433b-8775-48b247f356f0] I have attached the .cif file in question. I have not tried to systematically figure out what causes this discrepancy, I have really only noticed it here, but it is very annoying when trying to draw fold diagrams. Any idea what is going on? cheers Dr. Paul Kirchberger Assistant Professor Department of Microbiology and Molecular Genetics Oklahoma State University Stillwater, OK 74078 [cid:image001.png@01D77897.E8EBA0B0]




Attachments (6)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Change History (9)
by , 14 months ago
by , 14 months ago
| Attachment: | fold_err3063490_k141_167892_gp5_model_0.cif added |
|---|
Added by email2trac
comment:1 by , 14 months ago
| Component: | Unassigned → Structure Analysis |
|---|---|
| Owner: | set to |
| Platform: | → all |
| Project: | → ChimeraX |
| Status: | new → accepted |
comment:2 by , 14 months ago
| Resolution: | → not a bug |
|---|---|
| Status: | accepted → closed |
Hi Paul,
The .cif file you provided lacks information about secondary structure assignments, so ChimeraX computes them itself. If you use the "hbonds" command to show hydrogen bonds, you can see that as the segment in question bows out and away from the sheet, it loses hydrogen bonds. Nonetheless, you can make the short strand longer and recover the strand after the turn by using a less strict hydrogen-bond energy cutoff in the secondary-structure computation. By default the computation uses -0.5 kcal/mol. If you use -0.28 kcal/mol, you get closer to the PDB depiction. The command for that would be "dssp energy -0.28".
You can also force particular consecutive residues to be strand by selecting then and running these commands:
setattr sel r ss_type 2
setattr sel r ss_id 101
In the latter command use different ss_ids for different segments, and use high integers so that they don't conflict with automatic assignments, which will be low integers.
--Eric
Eric Pettersen
UCSF Computer Graphics Lab
comment:3 by , 14 months ago
Oh, thank you so much for the explanation!
Dr. Paul Kirchberger
Assistant Professor
Department of Microbiology and Molecular Genetics
Oklahoma State University
Stillwater, OK 74078
[cid:image001.png@01D77897.E8EBA0B0]
________________________________
From: ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu>
Sent: Thursday, June 5, 2025 3:29 PM
To: pett@cgl.ucsf.edu <pett@cgl.ucsf.edu>; Kirchberger, Paul <paul.kirchberger@okstate.edu>
Subject: Re: [ChimeraX] #17918: Difference in secondary structure display between Mol* 3D and ChimeraX
#17918: Difference in secondary structure display between Mol* 3D and ChimeraX
-----------------------------------------+--------------------
Reporter: paul.kirchberger@… | Owner: pett
Type: defect | Status: closed
Priority: normal | Milestone:
Component: Structure Analysis | Version:
Resolution: not a bug | Keywords:
Blocked By: | Blocking:
Notify when closed: | Platform: all
Project: ChimeraX |
-----------------------------------------+--------------------
Changes (by pett):
* resolution: => not a bug
* status: accepted => closed
Comment:
Hi Paul,
The .cif file you provided lacks information about secondary
structure assignments, so ChimeraX computes them itself. If you use the
"hbonds" command to show hydrogen bonds, you can see that as the segment
in question bows out and away from the sheet, it loses hydrogen bonds.
Nonetheless, you can make the short strand longer and recover the strand
after the turn by using a less strict hydrogen-bond energy cutoff in the
secondary-structure computation. By default the computation uses -0.5
kcal/mol. If you use -0.28 kcal/mol, you get closer to the PDB depiction.
The command for that would be "dssp energy -0.28".
You can also force particular consecutive residues to be strand by
selecting then and running these commands:
setattr sel r ss_type 2
setattr sel r ss_id 101
In the latter command use different ss_ids for different segments, and use
high integers so that they don't conflict with automatic assignments,
which will be low integers.
--Eric
Eric Pettersen
UCSF Computer Graphics Lab
--
Ticket URL: <https://nam04.safelinks.protection.outlook.com/?url=https%3A%2F%2Fwww.rbvi.ucsf.edu%2Ftrac%2FChimeraX%2Fticket%2F17918%23comment%3A2&data=05%7C02%7Cpaul.kirchberger%40okstate.edu%7C0948262c805945fd37e008dda46fac71%7C2a69c91de8494e34a230cdf8b27e1964%7C0%7C0%7C638847521778181114%7CUnknown%7CTWFpbGZsb3d8eyJFbXB0eU1hcGkiOnRydWUsIlYiOiIwLjAuMDAwMCIsIlAiOiJXaW4zMiIsIkFOIjoiTWFpbCIsIldUIjoyfQ%3D%3D%7C0%7C%7C%7C&sdata=sTQAGQZTVE0T3UiY0mozmnQ2EIo8rOQOPfj%2FrRNi6ks%3D&reserved=0<https://www.rbvi.ucsf.edu/trac/ChimeraX/ticket/17918#comment:2>>
ChimeraX <https://nam04.safelinks.protection.outlook.com/?url=https%3A%2F%2Fwww.rbvi.ucsf.edu%2Fchimerax%2F&data=05%7C02%7Cpaul.kirchberger%40okstate.edu%7C0948262c805945fd37e008dda46fac71%7C2a69c91de8494e34a230cdf8b27e1964%7C0%7C0%7C638847521778203384%7CUnknown%7CTWFpbGZsb3d8eyJFbXB0eU1hcGkiOnRydWUsIlYiOiIwLjAuMDAwMCIsIlAiOiJXaW4zMiIsIkFOIjoiTWFpbCIsIldUIjoyfQ%3D%3D%7C0%7C%7C%7C&sdata=SAFAyE9q8YOo4H1PMdpqmCqZswpZp1aShDGENYjBpck%3D&reserved=0<https://www.rbvi.ucsf.edu/chimerax/>>
ChimeraX Issue Tracker

Added by email2trac