Opened 2 years ago
Last modified 2 years ago
#14723 feedback defect
Problem with MSF alignment file
| Reported by: | Owned by: | Eric Pettersen | |
|---|---|---|---|
| Priority: | normal | Milestone: | |
| Component: | Sequence | Version: | |
| Keywords: | Cc: | ||
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
The following bug report has been submitted:
Platform: Windows-10-10.0.19045
ChimeraX Version: 1.7.1 (2024-01-23 01:58:08 UTC)
Description
Unable to open .msf file in sequence viewer
Log:
Startup Messages
---
note | available bundle cache has not been initialized yet
You can double click a model's Name or ID in the model panel to edit those
fields
UCSF ChimeraX version: 1.7.1 (2024-01-23)
© 2016-2023 Regents of the University of California. All rights reserved.
How to cite UCSF ChimeraX
> ui tool show "Show Sequence Viewer"
> show # target m
Expected a collection of one of 'atoms', 'bonds', 'cartoons', 'models',
'pbonds', 'pseudobonds', 'ribbons', or 'surfaces' or a keyword
> open "D:/Thesis/Data/Skanda/Sequence alignment/Sequences -
> Copy/muscle_aligned.msf"
Summary of feedback from opening D:/Thesis/Data/Skanda/Sequence
alignment/Sequences - Copy/muscle_aligned.msf
---
notes | Alignment identifier is muscle_aligned.msf
Showing conservation header ("seq_conservation" residue attribute) for
alignment muscle_aligned.msf
Opened 21 sequences from muscle_aligned.msf
> select
Nothing selected
> ui tool show "Show Sequence Viewer"
> open "D:\Thesis\Data\Skanda\Sequence alignment\Sequences -
> Copy\muscle_aligned.msf" format msf
Summary of feedback from opening D:\Thesis\Data\Skanda\Sequence
alignment\Sequences - Copy\muscle_aligned.msf
---
notes | Alignment identifier is muscle_aligned.msf
Showing conservation header ("seq_conservation" residue attribute) for
alignment muscle_aligned.msf
Opened 21 sequences from muscle_aligned.msf
> open "D:\Thesis\Data\Skanda\Sequence alignment\Sequences -
> Copy\muscle_aligned.msf" format msf
Summary of feedback from opening D:\Thesis\Data\Skanda\Sequence
alignment\Sequences - Copy\muscle_aligned.msf
---
notes | Destroying pre-existing alignment with identifier muscle_aligned.msf
Alignment identifier is muscle_aligned.msf
Showing conservation header ("seq_conservation" residue attribute) for
alignment muscle_aligned.msf
Opened 21 sequences from muscle_aligned.msf
> ~hbonds
> hbonds reveal true
Atom specifier selects no atoms
> interfaces ~solvent
No atoms specified
> color bynucleotide
> color bfactor
No atoms specified
> open "D:\Thesis\Data\Skanda\Sequence alignment\Sequences -
> Copy\muscle_aligned.msf" format msf
Summary of feedback from opening D:\Thesis\Data\Skanda\Sequence
alignment\Sequences - Copy\muscle_aligned.msf
---
notes | Destroying pre-existing alignment with identifier muscle_aligned.msf
Alignment identifier is muscle_aligned.msf
Showing conservation header ("seq_conservation" residue attribute) for
alignment muscle_aligned.msf
Opened 21 sequences from muscle_aligned.msf
> close #
Expected a models specifier or a keyword
> ui windowfill toggle
[Repeated 1 time(s)]
> ui tool show "Blast Protein"
> open "D:\Thesis\Data\Skanda\Sequence alignment\Sequences -
> Copy\muscle_aligned.msf" format msf
Summary of feedback from opening D:\Thesis\Data\Skanda\Sequence
alignment\Sequences - Copy\muscle_aligned.msf
---
notes | Destroying pre-existing alignment with identifier muscle_aligned.msf
Alignment identifier is muscle_aligned.msf
Showing conservation header ("seq_conservation" residue attribute) for
alignment muscle_aligned.msf
Opened 21 sequences from muscle_aligned.msf
> open "D:\Thesis\Data\Skanda\Sequence alignment\Sequences -
> Copy\muscle_aligned.msf" format msf
Summary of feedback from opening D:\Thesis\Data\Skanda\Sequence
alignment\Sequences - Copy\muscle_aligned.msf
---
notes | Destroying pre-existing alignment with identifier muscle_aligned.msf
Alignment identifier is muscle_aligned.msf
Showing conservation header ("seq_conservation" residue attribute) for
alignment muscle_aligned.msf
Opened 21 sequences from muscle_aligned.msf
> open "D:\Thesis\Data\Skanda\Sequence alignment\Sequences -
> Copy\muscle_aligned.msf" format fasta
No sequences found in FASTA file 'muscle_aligned.msf'!
> open "D:\Thesis\Data\Skanda\Sequence alignment\Sequences -
> Copy\muscle_aligned.msf" format afas
Invalid "format" argument: Should be one of 'aln', 'amber', 'amira', 'apbs',
'bild', 'brix', 'ccd', 'ccp4', 'cellpack', 'clustal', 'cmap', 'cmd', 'cod',
'collada', 'compiled python', 'corecif', 'cube', 'dcd', 'defattr', 'delphi',
'dicom', 'dock', 'dsn6', 'dv', 'emanhdf', 'fasta', 'fsc', 'gltf', 'gopenmol',
'gro', 'hdf', 'hssp', 'html', 'ihm', 'images', 'imagic', 'imod', 'imodmap',
'ims', 'iupac', 'macmolplt', 'markers', 'mmcif', 'mmtf', 'mol2', 'mole',
'mrc', 'msf', 'netcdfmap', 'nifti', 'nrrd', 'obj', 'pcod', 'pdb', 'pdbqt',
'pfam', 'photo', 'pif', 'pir', 'positions', 'priism', 'profec', 'pseudobonds',
'psf', 'python', 'rsf', 'schrodinger maestro', 'sdf', 'segger', 'session',
'sff', 'situs', 'smallcif', 'smiles', 'spider', 'stl', 'stockholm', 'storm',
'swc', 'swissdock', 'tom_em', 'trr', 'uhbd', 'uniprot', 'viperdb', 'vtk', 'web
fetch', 'xplor', 'xtc', or 'zdock'
> open "D:\Thesis\Data\Skanda\Sequence alignment\Sequences -
> Copy\muscle_aligned.msf" format aln
Cannot open files: Syntax error in ALN file 'muscle_aligned.msf': First non-
blank line does not start with 'CLUSTAL'
> open "D:\Thesis\Data\Skanda\Sequence alignment\Sequences -
> Copy\muscle_aligned.msf" format msn
Invalid "format" argument: Should be one of 'aln', 'amber', 'amira', 'apbs',
'bild', 'brix', 'ccd', 'ccp4', 'cellpack', 'clustal', 'cmap', 'cmd', 'cod',
'collada', 'compiled python', 'corecif', 'cube', 'dcd', 'defattr', 'delphi',
'dicom', 'dock', 'dsn6', 'dv', 'emanhdf', 'fasta', 'fsc', 'gltf', 'gopenmol',
'gro', 'hdf', 'hssp', 'html', 'ihm', 'images', 'imagic', 'imod', 'imodmap',
'ims', 'iupac', 'macmolplt', 'markers', 'mmcif', 'mmtf', 'mol2', 'mole',
'mrc', 'msf', 'netcdfmap', 'nifti', 'nrrd', 'obj', 'pcod', 'pdb', 'pdbqt',
'pfam', 'photo', 'pif', 'pir', 'positions', 'priism', 'profec', 'pseudobonds',
'psf', 'python', 'rsf', 'schrodinger maestro', 'sdf', 'segger', 'session',
'sff', 'situs', 'smallcif', 'smiles', 'spider', 'stl', 'stockholm', 'storm',
'swc', 'swissdock', 'tom_em', 'trr', 'uhbd', 'uniprot', 'viperdb', 'vtk', 'web
fetch', 'xplor', 'xtc', or 'zdock'
OpenGL version: 3.3.0 NVIDIA 546.26
OpenGL renderer: NVIDIA GeForce MX130/PCIe/SSE2
OpenGL vendor: NVIDIA Corporation
Python: 3.11.2
Locale: en_US.cp1252
Qt version: PyQt6 6.3.1, Qt 6.3.1
Qt runtime version: 6.3.2
Qt platform: windows
Manufacturer: HP
Model: HP Pavilion Laptop 15-cs1xxx
OS: Microsoft Windows 10 Home Single Language (Build 19045)
Memory: 8,471,760,896
MaxProcessMemory: 137,438,953,344
CPU: 8 Intel(R) Core(TM) i5-8265U CPU @ 1.60GHz
OSLanguage: en-US
Installed Packages:
alabaster: 0.7.16
appdirs: 1.4.4
asttokens: 2.4.1
Babel: 2.14.0
backcall: 0.2.0
beautifulsoup4: 4.11.2
blockdiag: 3.0.0
blosc2: 2.0.0
build: 0.10.0
certifi: 2023.11.17
cftime: 1.6.3
charset-normalizer: 3.3.2
ChimeraX-AddCharge: 1.5.13
ChimeraX-AddH: 2.2.5
ChimeraX-AlignmentAlgorithms: 2.0.1
ChimeraX-AlignmentHdrs: 3.4.1
ChimeraX-AlignmentMatrices: 2.1
ChimeraX-Alignments: 2.12.2
ChimeraX-AlphaFold: 1.0
ChimeraX-AltlocExplorer: 1.1.1
ChimeraX-AmberInfo: 1.0
ChimeraX-Arrays: 1.1
ChimeraX-Atomic: 1.49.1
ChimeraX-AtomicLibrary: 12.1.5
ChimeraX-AtomSearch: 2.0.1
ChimeraX-AxesPlanes: 2.3.2
ChimeraX-BasicActions: 1.1.2
ChimeraX-BILD: 1.0
ChimeraX-BlastProtein: 2.1.2
ChimeraX-BondRot: 2.0.4
ChimeraX-BugReporter: 1.0.1
ChimeraX-BuildStructure: 2.10.5
ChimeraX-Bumps: 1.0
ChimeraX-BundleBuilder: 1.2.2
ChimeraX-ButtonPanel: 1.0.1
ChimeraX-CageBuilder: 1.0.1
ChimeraX-CellPack: 1.0
ChimeraX-Centroids: 1.3.2
ChimeraX-ChangeChains: 1.1
ChimeraX-CheckWaters: 1.3.2
ChimeraX-ChemGroup: 2.0.1
ChimeraX-Clashes: 2.2.4
ChimeraX-ColorActions: 1.0.3
ChimeraX-ColorGlobe: 1.0
ChimeraX-ColorKey: 1.5.5
ChimeraX-CommandLine: 1.2.5
ChimeraX-ConnectStructure: 2.0.1
ChimeraX-Contacts: 1.0.1
ChimeraX-Core: 1.7.1
ChimeraX-CoreFormats: 1.2
ChimeraX-coulombic: 1.4.2
ChimeraX-Crosslinks: 1.0
ChimeraX-Crystal: 1.0
ChimeraX-CrystalContacts: 1.0.1
ChimeraX-DataFormats: 1.2.3
ChimeraX-Dicom: 1.2
ChimeraX-DistMonitor: 1.4
ChimeraX-DockPrep: 1.1.3
ChimeraX-Dssp: 2.0
ChimeraX-EMDB-SFF: 1.0
ChimeraX-ESMFold: 1.0
ChimeraX-FileHistory: 1.0.1
ChimeraX-FunctionKey: 1.0.1
ChimeraX-Geometry: 1.3
ChimeraX-gltf: 1.0
ChimeraX-Graphics: 1.1.1
ChimeraX-Hbonds: 2.4
ChimeraX-Help: 1.2.2
ChimeraX-HKCage: 1.3
ChimeraX-IHM: 1.1
ChimeraX-ImageFormats: 1.2
ChimeraX-IMOD: 1.0
ChimeraX-IO: 1.0.1
ChimeraX-ItemsInspection: 1.0.1
ChimeraX-IUPAC: 1.0
ChimeraX-Label: 1.1.8
ChimeraX-ListInfo: 1.2.2
ChimeraX-Log: 1.1.6
ChimeraX-LookingGlass: 1.1
ChimeraX-Maestro: 1.9.1
ChimeraX-Map: 1.1.4
ChimeraX-MapData: 2.0
ChimeraX-MapEraser: 1.0.1
ChimeraX-MapFilter: 2.0.1
ChimeraX-MapFit: 2.0
ChimeraX-MapSeries: 2.1.1
ChimeraX-Markers: 1.0.1
ChimeraX-Mask: 1.0.2
ChimeraX-MatchMaker: 2.1.2
ChimeraX-MCopy: 1.0
ChimeraX-MDcrds: 2.6.1
ChimeraX-MedicalToolbar: 1.0.2
ChimeraX-Meeting: 1.0.1
ChimeraX-MLP: 1.1.1
ChimeraX-mmCIF: 2.12.1
ChimeraX-MMTF: 2.2
ChimeraX-Modeller: 1.5.14
ChimeraX-ModelPanel: 1.4
ChimeraX-ModelSeries: 1.0.1
ChimeraX-Mol2: 2.0.3
ChimeraX-Mole: 1.0
ChimeraX-Morph: 1.0.2
ChimeraX-MouseModes: 1.2
ChimeraX-Movie: 1.0
ChimeraX-Neuron: 1.0
ChimeraX-Nifti: 1.1
ChimeraX-NRRD: 1.1
ChimeraX-Nucleotides: 2.0.3
ChimeraX-OpenCommand: 1.13.1
ChimeraX-PDB: 2.7.3
ChimeraX-PDBBio: 1.0.1
ChimeraX-PDBLibrary: 1.0.4
ChimeraX-PDBMatrices: 1.0
ChimeraX-PickBlobs: 1.0.1
ChimeraX-Positions: 1.0
ChimeraX-PresetMgr: 1.1
ChimeraX-PubChem: 2.1
ChimeraX-ReadPbonds: 1.0.1
ChimeraX-Registration: 1.1.2
ChimeraX-RemoteControl: 1.0
ChimeraX-RenderByAttr: 1.1
ChimeraX-RenumberResidues: 1.1
ChimeraX-ResidueFit: 1.0.1
ChimeraX-RestServer: 1.2
ChimeraX-RNALayout: 1.0
ChimeraX-RotamerLibMgr: 4.0
ChimeraX-RotamerLibsDunbrack: 2.0
ChimeraX-RotamerLibsDynameomics: 2.0
ChimeraX-RotamerLibsRichardson: 2.0
ChimeraX-SaveCommand: 1.5.1
ChimeraX-SchemeMgr: 1.0
ChimeraX-SDF: 2.0.2
ChimeraX-Segger: 1.0
ChimeraX-Segment: 1.0.1
ChimeraX-SelInspector: 1.0
ChimeraX-SeqView: 2.11
ChimeraX-Shape: 1.0.1
ChimeraX-Shell: 1.0.1
ChimeraX-Shortcuts: 1.1.1
ChimeraX-ShowSequences: 1.0.2
ChimeraX-SideView: 1.0.1
ChimeraX-Smiles: 2.1.2
ChimeraX-SmoothLines: 1.0
ChimeraX-SpaceNavigator: 1.0
ChimeraX-StdCommands: 1.12.4
ChimeraX-STL: 1.0.1
ChimeraX-Storm: 1.0
ChimeraX-StructMeasure: 1.1.2
ChimeraX-Struts: 1.0.1
ChimeraX-Surface: 1.0.1
ChimeraX-SwapAA: 2.0.1
ChimeraX-SwapRes: 2.2.2
ChimeraX-TapeMeasure: 1.0
ChimeraX-TaskManager: 1.0
ChimeraX-Test: 1.0
ChimeraX-Toolbar: 1.1.2
ChimeraX-ToolshedUtils: 1.2.4
ChimeraX-Topography: 1.0
ChimeraX-ToQuest: 1.0
ChimeraX-Tug: 1.0.1
ChimeraX-UI: 1.33.3
ChimeraX-uniprot: 2.3
ChimeraX-UnitCell: 1.0.1
ChimeraX-ViewDockX: 1.3.2
ChimeraX-VIPERdb: 1.0
ChimeraX-Vive: 1.1
ChimeraX-VolumeMenu: 1.0.1
ChimeraX-vrml: 1.0
ChimeraX-VTK: 1.0
ChimeraX-WavefrontOBJ: 1.0
ChimeraX-WebCam: 1.0.2
ChimeraX-WebServices: 1.1.3
ChimeraX-Zone: 1.0.1
colorama: 0.4.6
comm: 0.2.1
comtypes: 1.1.14
contourpy: 1.2.0
cxservices: 1.2.2
cycler: 0.12.1
Cython: 0.29.33
debugpy: 1.8.0
decorator: 5.1.1
docutils: 0.19
executing: 2.0.1
filelock: 3.9.0
fonttools: 4.47.2
funcparserlib: 2.0.0a0
glfw: 2.6.4
grako: 3.16.5
h5py: 3.10.0
html2text: 2020.1.16
idna: 3.6
ihm: 0.38
imagecodecs: 2023.9.18
imagesize: 1.4.1
ipykernel: 6.23.2
ipython: 8.14.0
ipython-genutils: 0.2.0
ipywidgets: 8.1.1
jedi: 0.18.2
Jinja2: 3.1.2
jupyter-client: 8.2.0
jupyter-core: 5.7.1
jupyterlab-widgets: 3.0.9
kiwisolver: 1.4.5
line-profiler: 4.0.2
lxml: 4.9.2
lz4: 4.3.2
MarkupSafe: 2.1.4
matplotlib: 3.7.2
matplotlib-inline: 0.1.6
msgpack: 1.0.4
nest-asyncio: 1.6.0
netCDF4: 1.6.2
networkx: 3.1
nibabel: 5.0.1
nptyping: 2.5.0
numexpr: 2.8.8
numpy: 1.25.1
openvr: 1.23.701
packaging: 23.2
ParmEd: 3.4.3
parso: 0.8.3
pep517: 0.13.0
pickleshare: 0.7.5
pillow: 10.2.0
pip: 23.0
pkginfo: 1.9.6
platformdirs: 4.1.0
prompt-toolkit: 3.0.43
psutil: 5.9.5
pure-eval: 0.2.2
py-cpuinfo: 9.0.0
pycollada: 0.7.2
pydicom: 2.3.0
Pygments: 2.16.1
pynrrd: 1.0.0
PyOpenGL: 3.1.7
PyOpenGL-accelerate: 3.1.7
pyopenxr: 1.0.2801
pyparsing: 3.0.9
pyproject-hooks: 1.0.0
PyQt6-commercial: 6.3.1
PyQt6-Qt6: 6.3.2
PyQt6-sip: 13.4.0
PyQt6-WebEngine-commercial: 6.3.1
PyQt6-WebEngine-Qt6: 6.3.2
python-dateutil: 2.8.2
pytz: 2023.3.post1
pywin32: 305
pyzmq: 25.1.2
qtconsole: 5.4.3
QtPy: 2.4.1
RandomWords: 0.4.0
requests: 2.31.0
scipy: 1.11.1
setuptools: 67.4.0
sfftk-rw: 0.7.3
six: 1.16.0
snowballstemmer: 2.2.0
sortedcontainers: 2.4.0
soupsieve: 2.5
sphinx: 6.1.3
sphinx-autodoc-typehints: 1.22
sphinxcontrib-applehelp: 1.0.8
sphinxcontrib-blockdiag: 3.0.0
sphinxcontrib-devhelp: 1.0.6
sphinxcontrib-htmlhelp: 2.0.5
sphinxcontrib-jsmath: 1.0.1
sphinxcontrib-qthelp: 1.0.7
sphinxcontrib-serializinghtml: 1.1.10
stack-data: 0.6.3
superqt: 0.5.0
tables: 3.8.0
tcia-utils: 1.5.1
tifffile: 2023.7.18
tinyarray: 1.2.4
tomli: 2.0.1
tornado: 6.4
traitlets: 5.9.0
typing-extensions: 4.9.0
tzdata: 2023.4
urllib3: 2.1.0
wcwidth: 0.2.13
webcolors: 1.12
wheel: 0.38.4
wheel-filename: 1.4.1
widgetsnbextension: 4.0.9
WMI: 1.5.1
Attachments (1)
{kind=link}
Change History (5)
comment:1 by , 2 years ago
| Component: | Unassigned → Sequence |
|---|---|
| Owner: | set to |
| Platform: | → all |
| Project: | → ChimeraX |
| Status: | new → accepted |
| Summary: | ChimeraX bug report submission → Problem with MSF alignment file |
comment:2 by , 2 years ago
| Status: | accepted → feedback |
|---|
comment:3 by , 2 years ago
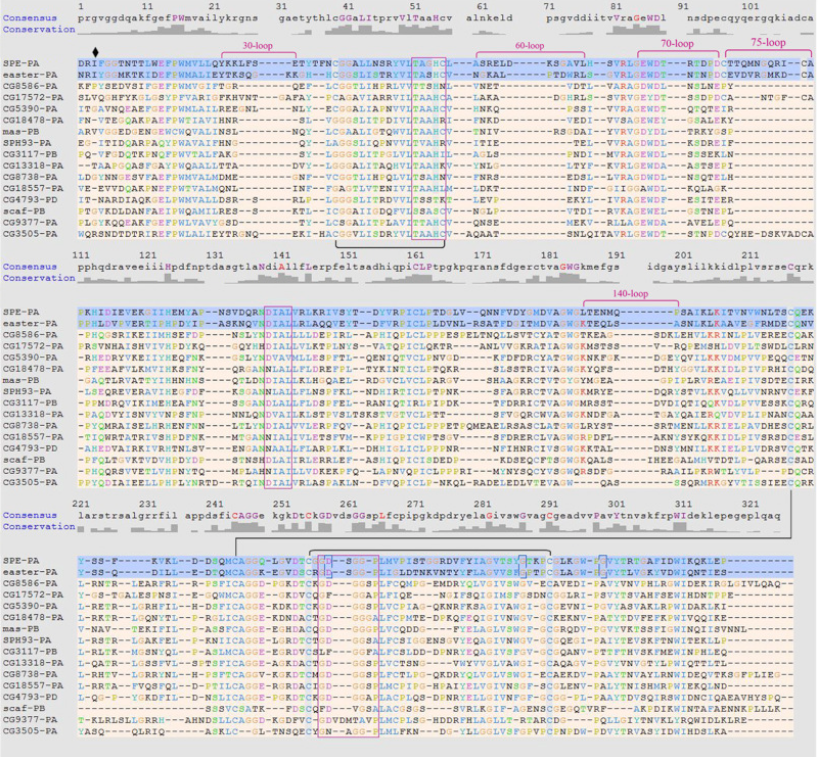
Hi Eric, My problem is that I was unable to open the .msf file using the ChimeraX sequence viewer. I wanted to see the conserved residues and consensus sequence, but despite multiple attempts following the tutorial, I was unable to complete the action. The sequence viewer was open, but I was unable to select the sequences on the .msf file that I wanted to view. I essentially wanted an output like the attached image. Please let me know if I'm doing something wrong. Thanks, Sanjana [image: image.png] On Thu, Mar 7, 2024 at 10:29 PM ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote: > > > > > >

comment:4 by , 2 years ago
Hi Sanjana,
The conservation header should be shown by default initially. To show the consensus header, right click on the sequence viewer and in the resulting popup menu select Headers→Consensus. Let me know if you have further questions.
--Eric
Note:
See TracTickets
for help on using tickets.
Hi Sanjana,
--Eric