#10447 closed enhancement (fixed)
Allow display NMR constraints as pseudobonds for PDB structures
| Reported by: | Tom Goddard | Owned by: | Tom Goddard |
|---|---|---|---|
| Priority: | moderate | Milestone: | |
| Component: | Structure Analysis | Version: | |
| Keywords: | Cc: | Elaine Meng, Eric Pettersen | |
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
Attachments (1)
{kind=link}
{kind=link}
Change History (6)
comment:1 by , 3 years ago
| Resolution: | → fixed |
|---|---|
| Status: | assigned → closed |
comment:3 by , 3 years ago
Yeah I considered nmrsatisfied, nmrlong and was undecided by leaned slightly towards the easier to type. It can of course be changed. Let's see what Elaine thinks.
comment:4 by , 3 years ago
I would vote for nmr-long and nmr-satisfied as smushing it together makes it harder to read, but would settle for nmrlong and nmrsatisfied if you prefer. Otherwise they seem too generic, in case we (or some plugin developers) eventually want to have other types of constraints that might also also be satisfied or too-long. We do have others with hyphens already, e.g. aromatic-ring, template-mismatch, min-backbone: https://www.rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#builtin
comment:5 by , 3 years ago
Ok, I'll change the names to nmr-satisfied and nmr-long tomorrow after the class.
Note:
See TracTickets
for help on using tickets.
Done.
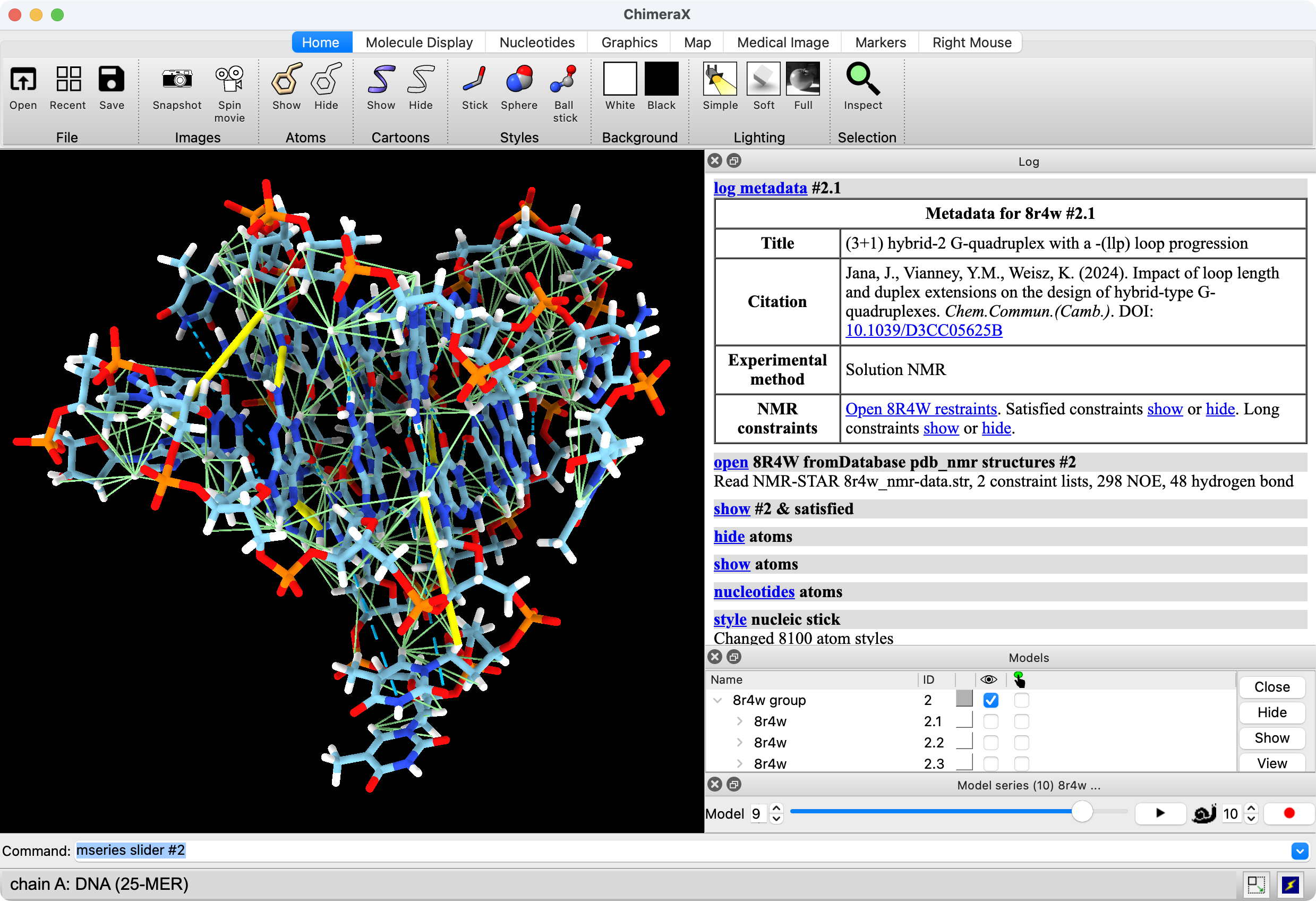
Can now open NMR-STAR files (suffix .str) and can fetch them for PDB entries. For example,
The .str files can be downloaded from the PDB entry pages in their Download menu, the entry that says "Combined NMR data (NMR-STAR) (.gz)".
The "structures" option should be specified when opening NMR-STAR data to tell which atomic structures the constraints should be shown on as pseudobonds. If the structures option is omitted then opening the NMR-STAR file only reports how many constraints were found in the file.
Multiple sets of distance constraints can be in a single NMR-STAR file, and each set has a type such as "NOE" or "hydrogen bond". By default each shown as a separate pseudobond group with the type in the name ("NOE constraints", "hydrogen bond constraints"). The pseudobond groups are children of the structures they are applied to. To load just one type of constraint use the "type" option of the open command
Each distance constraint has a maximum distance. If the distance in the structure is less than the maximum it is satisfied, otherwise it is long. The satisfied constraint pseudobonds are colored light green (160,255,160) and have radius 0.05 and the long ones are colored yellow (255,255,0) with radius 0.2. All of them are solid (no dashes). Those values can be changed, for example,
There are two selectors to help hide and show satisfied or long constraints, named "satisfied" and "long"
The constraints are saved in sessions including their maximum distance.
When a PDB NMR entry is opened which has NMR-STAR data (only found in newer entries) the "more info" table in the Log gives a link to open the constraints, and also hide and show links for satisfied and long constraints to help users learn those selectors.