Opened 6 years ago
Last modified 16 months ago
#4176 closed defect
ChimeraX bug report submission — at Initial Version
| Reported by: | Tristan Croll | Owned by: | |
|---|---|---|---|
| Priority: | normal | Milestone: | |
| Component: | Input/Output | Version: | |
| Keywords: | Cc: | Eric Pettersen | |
| Blocked By: | Blocking: | ||
| Notify when closed: | Platform: | all | |
| Project: | ChimeraX |
Description
The following bug report has been submitted:
Platform: Linux-3.10.0-1127.19.1.el7.x86_64-x86_64-with-centos-7.8.2003-Core
ChimeraX Version: 1.1 (2020-09-09 22:22:27 UTC)
Description
I have an ISOLDE session containing a very large, complex model. Everything in the session itself is well-behaved, but when I save to mmCIF and reopen five residues are duplicated. One of these is a protein residue (somehow ended up appearing twice in its `Chain` object, no idea how - easy enough to fix). The other four are two pairs of sugars, each internal to an O-linked glycan (like in the attached image). Much less sure how to fix that, other than to delete the duplicate residues in the reopened mmCIF. Any clues?
Log:
UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Mon Jan 25 22:36:36 2021UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Sat Jan 23 15:29:45 2021UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Wed Jan 20 20:19:45 2021UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Wed Jan 20 18:04:18 2021UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working_fme.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Failed to restore chiral restraints. This is usually not a problem - they will
be automatically regenerated on first simulation.
Log from Tue Jan 19 20:41:55 2021UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working_fme.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Failed to restore chiral restraints. This is usually not a problem - they will
be automatically regenerated on first simulation.
Log from Tue Jan 19 19:42:56 2021UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Tue Jan 19 15:18:30 2021UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Mon Jan 18 21:58:27 2021UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Thu Jan 14 20:29:04 2021UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Thu Jan 14 18:05:53 2021UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open model1a_2020_11_04.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Wed Nov 4 20:47:05 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open model1a_2020_11_02.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Mon Nov 2 17:41:56 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Sat Oct 31 21:36:29 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1a/reassigning_ligands.cxs
> format session
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Fri Oct 30 09:34:25 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1a/reassigning_ligands.cxs
> format session
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Fri Oct 30 09:16:38 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Thu Oct 29 16:35:33 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Wed Oct 28 21:12:33 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Tue Oct 27 22:03:57 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Fri Oct 23 21:33:44 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Fri Oct 23 12:38:35 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Thu Oct 22 21:38:47 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working_3.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Thu Oct 22 17:00:13 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1b/reopened.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Thu Oct 22 16:23:14 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open working_2.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Thu Oct 22 16:02:14 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1b/working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Thu Oct 22 13:56:05 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open ../old_model_in_map_1b.cxs
Log from Thu Oct 22 06:57:21 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open model1b_working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Wed Oct 21 20:17:18 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open model1b_working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Wed Oct 21 20:13:59 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
> open
> /run/media/tic20/storage/structure_dump/pu_qian/2020_10_new_maps/model1b_working.cxs
restore_snapshot for "RotamerRestraintMgr" returned None
Log from Wed Oct 21 19:55:36 2020UCSF ChimeraX version: 1.1 (2020-09-09)
© 2016-2020 Regents of the University of California. All rights reserved.
How to cite UCSF ChimeraX
> open /run/media/tic20/storage/structure_dump/pu_qian/2020_10_new_maps/rc-
> dlh1-model1b_rearranged.pdb format pdb
Chain information for rc-dlh1-model1b_rearranged.pdb #1
---
Chain | Description
AA AB AC AD AE AF AG AH AI AJ AK AL AM AN AO AP AQ AR AS AT AU AV AW AX | No
description available
Aa Ab Ac Ad Ae Af Ah Ai Aj Ak Al Am An Ao Ap | No description available
Ag | No description available
B | No description available
BA BB BC BD BE BF BG BH BI BJ BK BL BM BN BO BP BQ BR BS BT BU BV BW BX Ba Bb
Bc Bd Be Bf Bg Bh Bi Bj Bk Bl Bm Bn Bo Bp | No description available
C | No description available
C1 | No description available
H1 | No description available
H2 | No description available
L | No description available
M | No description available
> open
> /run/media/tic20/storage/structure_dump/pu_qian/2020_10_new_maps/class1b_26A.mrc
Opened class1b_26A.mrc, grid size 400,400,400, pixel 0.999, shown at level
0.0224, step 2, values float32
> clipper associate #2 toModel #1
Chain information for rc-dlh1-model1b_rearranged.pdb
---
Chain | Description
1.2/AA 1.2/AB 1.2/AC 1.2/AD 1.2/AE 1.2/AF 1.2/AG 1.2/AH 1.2/AI 1.2/AJ 1.2/AK
1.2/AL 1.2/AM 1.2/AN 1.2/AO 1.2/AP 1.2/AQ 1.2/AR 1.2/AS 1.2/AT 1.2/AU 1.2/AV
1.2/AW 1.2/AX | No description available
1.2/Aa 1.2/Ab 1.2/Ac 1.2/Ad 1.2/Ae 1.2/Af 1.2/Ah 1.2/Ai 1.2/Aj 1.2/Ak 1.2/Al
1.2/Am 1.2/An 1.2/Ao 1.2/Ap | No description available
1.2/Ag | No description available
1.2/B | No description available
1.2/BA 1.2/BB 1.2/BC 1.2/BD 1.2/BE 1.2/BF 1.2/BG 1.2/BH 1.2/BI 1.2/BJ 1.2/BK
1.2/BL 1.2/BM 1.2/BN 1.2/BO 1.2/BP 1.2/BQ 1.2/BR 1.2/BS 1.2/BT 1.2/BU 1.2/BV
1.2/BW 1.2/BX 1.2/Ba 1.2/Bb 1.2/Bc 1.2/Bd 1.2/Be 1.2/Bf 1.2/Bg 1.2/Bh 1.2/Bi
1.2/Bj 1.2/Bk 1.2/Bl 1.2/Bm 1.2/Bn 1.2/Bo 1.2/Bp | No description available
1.2/C | No description available
1.2/C1 | No description available
1.2/H1 | No description available
1.2/H2 | No description available
1.2/L | No description available
1.2/M | No description available
> delete :GPC@HO45
> ui tool show Shell
/opt/UCSF/ChimeraX/lib/python3.7/site-packages/IPython/core/history.py:226:
UserWarning: IPython History requires SQLite, your history will not be saved
warn("IPython History requires SQLite, your history will not be saved")
> select ~protein
10060 atoms, 10377 bonds, 352 pseudobonds, 135 residues, 8 models selected
> ui tool show ISOLDE
> set selectionWidth 4
Done loading forcefield
> select ~protein
10060 atoms, 10377 bonds, 352 pseudobonds, 135 residues, 13 models selected
> addh
Summary of feedback from adding hydrogens to rc-dlh1-model1b_rearranged.pdb
#1.2
---
warning | Skipped 8 atom(s) with bad connectivities; see log for details
notes | No usable SEQRES records for rc-dlh1-model1b_rearranged.pdb (#1.2)
chain AA; guessing termini instead
No usable SEQRES records for rc-dlh1-model1b_rearranged.pdb (#1.2) chain AB;
guessing termini instead
No usable SEQRES records for rc-dlh1-model1b_rearranged.pdb (#1.2) chain AC;
guessing termini instead
No usable SEQRES records for rc-dlh1-model1b_rearranged.pdb (#1.2) chain AD;
guessing termini instead
No usable SEQRES records for rc-dlh1-model1b_rearranged.pdb (#1.2) chain AE;
guessing termini instead
82 messages similar to the above omitted
Chain-initial residues that are actual N termini: /AA HIS 2, /AB HIS 2, /AC
HIS 2, /AD HIS 2, /AE HIS 2, /AF HIS 2, /AG HIS 2, /AH HIS 2, /AI HIS 2, /AJ
HIS 2, /AK HIS 2, /AL HIS 2, /AM HIS 2, /AN HIS 2, /AO HIS 2, /AP HIS 2, /AQ
HIS 2, /AR HIS 2, /AS HIS 2, /AT HIS 2, /AU HIS 2, /AV HIS 2, /AW HIS 2, /AX
HIS 2, /Aa MET 1, /Ab MET 1, /Ac MET 1, /Ad MET 1, /Ae MET 1, /Af MET 1, /Ag
MET 1, /Ah MET 1, /Ai MET 1, /Aj MET 1, /Ak MET 1, /Al MET 1, /Am MET 1, /An
MET 1, /Ao MET 1, /Ap MET 1, /B PRO 25, /BA MET 1, /BB MET 1, /BC MET 1, /BD
MET 1, /BE MET 1, /BF MET 1, /BG MET 1, /BH MET 1, /BI MET 1, /BJ MET 1, /BK
MET 1, /BL MET 1, /BM MET 1, /BN MET 1, /BO MET 1, /BP MET 1, /BQ MET 1, /BR
MET 1, /BS MET 1, /BT MET 1, /BU MET 1, /BV MET 1, /BW MET 1, /BX MET 1, /Ba
MET 1, /Bb MET 1, /Bc MET 1, /Bd MET 1, /Be MET 1, /Bf MET 1, /Bg MET 1, /Bh
MET 1, /Bi MET 1, /Bj MET 1, /Bk MET 1, /Bl MET 1, /Bm MET 1, /Bn MET 1, /Bo
MET 1, /Bp MET 1, /C ALA 15, /H1 MET 1, /H2 SER 1, /L ALA 1, /M MET 1
Chain-initial residues that are not actual N termini: /C1 GLN 1, /H2 ARG 38
Chain-final residues that are actual C termini: /BA PHE 38, /BB PHE 38, /BC
PHE 38, /BD PHE 38, /BE PHE 38, /BF PHE 38, /BG PHE 38, /BH PHE 38, /BI PHE
38, /BJ PHE 38, /BK PHE 38, /BL PHE 38, /BM PHE 38, /BN PHE 38, /BO PHE 38,
/BP PHE 38, /BQ PHE 38, /BR PHE 38, /BS PHE 38, /BT PHE 38, /BU PHE 38, /BV
PHE 38, /BW PHE 38, /BX PHE 38, /Ba PHE 38, /Bb PHE 38, /Bc PHE 38, /Bd PHE
38, /Be PHE 38, /Bf PHE 38, /Bg PHE 38, /Bh PHE 38, /Bi PHE 38, /Bj PHE 38,
/Bk PHE 38, /Bl PHE 38, /Bm PHE 38, /Bn PHE 38, /Bo PHE 38, /Bp PHE 38
Chain-final residues that are not actual C termini: /AA PRO 48, /AB PRO 48,
/AC PRO 48, /AD PRO 48, /AE PRO 48, /AF PRO 48, /AG PRO 48, /AH PRO 48, /AI
PRO 48, /AJ PRO 48, /AK PRO 48, /AL PRO 48, /AM PRO 48, /AN PRO 48, /AO PRO
48, /AP PRO 48, /AQ PRO 48, /AR PRO 48, /AS PRO 48, /AT PRO 48, /AU PRO 48,
/AV PRO 48, /AW PRO 48, /AX PRO 48, /Aa ALA 60, /Ab ALA 60, /Ac ALA 60, /Ad
ALA 60, /Ae ALA 60, /Af ALA 60, /Ag LEU 67, /Ah ALA 60, /Ai ALA 60, /Aj ALA
60, /Ak ALA 60, /Al ALA 60, /Am ALA 60, /An ALA 60, /Ao ALA 60, /Ap ALA 60, /B
ILE 123, /C ILE 313, /C1 LEU 92, /H1 HIS 67, /H2 ILE 181, /H2 ARG 36, /L TRP
272, /M TYR 324
Skipping possible acceptor with bad geometry: /C ARG 302 O
Wrong number of grandchild atoms for phi/psi acceptor /C ARG 302 O
Skipping possible acceptor with bad geometry: /C ARG 302 OXT
Wrong number of grandchild atoms for phi/psi acceptor /C ARG 302 OXT
Skipping possible acceptor with bad geometry: /C ARG 302 OXT
Wrong number of grandchild atoms for phi/psi acceptor /C ARG 302 OXT
Skipping possible acceptor with bad geometry: /C ARG 302 O
Wrong number of grandchild atoms for phi/psi acceptor /C ARG 302 O
Skipping possible acceptor with bad geometry: /C ARG 302 O
Wrong number of grandchild atoms for phi/psi acceptor /C ARG 302 O
3 messages similar to the above omitted
4746 hydrogen bonds
Adding 'H' to /H2 ARG 38
/AA PRO 48 is not terminus, removing H atom from 'C'
/AB PRO 48 is not terminus, removing H atom from 'C'
/AC PRO 48 is not terminus, removing H atom from 'C'
/AD PRO 48 is not terminus, removing H atom from 'C'
/AE PRO 48 is not terminus, removing H atom from 'C'
42 messages similar to the above omitted
44090 hydrogens added
> save model1b_working.cxs
opened ChimeraX session
> select up
101 atoms, 100 bonds, 1 residue, 1 model selected
> show sel
> select clear
> style sel ball
Changed 100 atom styles
> select :GPC
4013 atoms, 3973 bonds, 40 residues, 1 model selected
> show sel
> select :GPC@HC19
40 atoms, 40 residues, 1 model selected
> select :GPC
4013 atoms, 3973 bonds, 40 residues, 1 model selected
> delete sel
MD template USER_BCL for residue BCL L604 contains extra atoms that are not in
a coordinate template, and are not directly connected to existing atoms. Since
MD templates do not explicitly provide geometry,these atoms will not be built.
> select sel&@CBD
1 atom, 1 residue, 1 model selected
> style sel sphere
Changed 1 atom style
> style sel stick
Changed 1 atom style
> select up
139 atoms, 147 bonds, 1 residue, 1 model selected
Deleted the following atoms from residue BCL L604: HHC1, HHD1
Deleted the following atoms from residue BCL L602: HHD2, HHC2, HHD1, HHC1
Deleted the following atoms from residue BPH L606: H12, H92, HHD1, H41, H93,
H91, H52, H11, HHD2, H43, H61, H51, H62, H42, H71, H72
> save model1b_working.cxs
opened ChimeraX session
Deleted the following atoms from residue MQ8 M405: H272, H9, H502, H122, C49,
C46, H172, H503, C50, H461, H471, H222, H11, H422, H492, H6, H472, C48, H462,
H7, H501, C47, H8, H491, H372, H322
Deleted the following atoms from residue BCL M601: HHD2, HHD1
Deleted the following atoms from residue BCL M603: HHD1, HHD2, HHC1, HHC2
Deleted the following atoms from residue BPH M605: H11, H62, H72, H42, H12,
H91, H52, H71, HHD2, H93, H51, H92, HHD1, H61, H43, H41
Residue RCC M701 has only 0 connected atoms in common with template GPC. At
least 3 matching atoms are needed.
> save model1b_working.cxs
> delete sel
> save model1b_working.cxs
opened ChimeraX session
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/RealSpaceRefine_44/working_noh_real_space_refined.pdb
Summary of feedback from opening
/run/media/tic20/storage/structure_dump/collaboration/pu_qian/RealSpaceRefine_44/working_noh_real_space_refined.pdb
---
warnings | Ignored bad PDB record found on line 317
LINK NE2 HIS AA 29 MG BCL AA 57
Ignored bad PDB record found on line 318
LINK NE2 HIS AB 29 MG BCL AB 57
Ignored bad PDB record found on line 319
LINK NE2 HIS AC 29 MG BCL AC 57
Ignored bad PDB record found on line 320
LINK NE2 HIS AD 29 MG BCL AD 57
Ignored bad PDB record found on line 321
LINK NE2 HIS AE 29 MG BCL AE 57
75 messages similar to the above omitted
Start residue of secondary structure not found: HELIX 1 1 PROAA 10 PHEAA 36 1
26
Start residue of secondary structure not found: HELIX 2 2 ALAAA 40 LYSAA 45 1
5
Start residue of secondary structure not found: HELIX 3 3 ARGAB 3 TRPAB 5 1 2
Start residue of secondary structure not found: HELIX 4 4 PROAB 10 GLNAB 35 1
25
Start residue of secondary structure not found: HELIX 5 5 ALAAB 40 LEUAB 42 1
2
207 messages similar to the above omitted
Chain information for working_noh_real_space_refined.pdb #2
---
Chain | Description
AA AB AC AD AE AF AG AH AI AJ AK AL AM AN AO AP AQ AR AS AT AU AV AW AX | No
description available
BA BC BF BG BH BJ BK BL BM BN BO BP BQ BR BS BT BU BX ba bb bc bd be bf bg bh
bi bj bk bl bm bo bp | No description available
BB BD BE BI BV BW bn | No description available
C | No description available
H1 | No description available
H2 | No description available
L | No description available
M | No description available
UA | No description available
UB | No description available
UC | No description available
aa | No description available
ab ac ad ae af ag ah ai aj ak al am an ao ap | No description available
> matchmaker #2 to #1
Parameters
---
Chain pairing | bb
Alignment algorithm | Needleman-Wunsch
Similarity matrix | BLOSUM-62
SS fraction | 0.3
Gap open (HH/SS/other) | 18/18/6
Gap extend | 1
SS matrix | | | H | S | O
---|---|---|---
H | 6 | -9 | -6
S | | 6 | -6
O | | | 4
Iteration cutoff | 2
Matchmaker rc-dlh1-model1b_rearranged.pdb, chain M (#1.2) with
working_noh_real_space_refined.pdb, chain M (#2), sequence alignment score =
1582
RMSD between 295 pruned atom pairs is 0.684 angstroms; (across all 324 pairs:
1.723)
> delete #2/UA,UB,UC
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/class1b_26A.mrc
Opened class1b_26A.mrc, grid size 400,400,400, pixel 0.999, shown at level
0.0224, step 2, values float32
> clipper associate #3 toModel #2
Chain information for working_noh_real_space_refined.pdb
---
Chain | Description
2.2/AA 2.2/AB 2.2/AC 2.2/AD 2.2/AE 2.2/AF 2.2/AG 2.2/AH 2.2/AI 2.2/AJ 2.2/AK
2.2/AL 2.2/AM 2.2/AN 2.2/AO 2.2/AP 2.2/AQ 2.2/AR 2.2/AS 2.2/AT 2.2/AU 2.2/AV
2.2/AW 2.2/AX | No description available
2.2/BA 2.2/BC 2.2/BF 2.2/BG 2.2/BH 2.2/BJ 2.2/BK 2.2/BL 2.2/BM 2.2/BN 2.2/BO
2.2/BP 2.2/BQ 2.2/BR 2.2/BS 2.2/BT 2.2/BU 2.2/BX 2.2/ba 2.2/bb 2.2/bc 2.2/bd
2.2/be 2.2/bf 2.2/bg 2.2/bh 2.2/bi 2.2/bj 2.2/bk 2.2/bl 2.2/bm 2.2/bo 2.2/bp |
No description available
2.2/BB 2.2/BD 2.2/BE 2.2/BI 2.2/BV 2.2/BW 2.2/bn | No description available
2.2/C | No description available
2.2/H1 | No description available
2.2/H2 | No description available
2.2/L | No description available
2.2/M | No description available
2.2/aa | No description available
2.2/ab 2.2/ac 2.2/ad 2.2/ae 2.2/af 2.2/ag 2.2/ah 2.2/ai 2.2/aj 2.2/ak 2.2/al
2.2/am 2.2/an 2.2/ao 2.2/ap | No description available
> select #2:GPC
1800 atoms, 1760 bonds, 40 residues, 1 model selected
> select #2:RCC
Nothing selected
> select #1:RCC
100 atoms, 99 bonds, 1 residue, 1 model selected
> select #2:GPC
1800 atoms, 1760 bonds, 40 residues, 1 model selected
> select #1:GPC
4000 atoms, 3960 bonds, 40 residues, 1 model selected
> select #2:GPC
1800 atoms, 1760 bonds, 40 residues, 1 model selected
> delete sel
> ui tool show Shell
/opt/UCSF/ChimeraX/lib/python3.7/site-packages/IPython/core/history.py:226:
UserWarning: IPython History requires SQLite, your history will not be saved
warn("IPython History requires SQLite, your history will not be saved")
> delete #2:GP1
> select #1:RCC
100 atoms, 99 bonds, 1 residue, 1 model selected
> select #1:RCC,GCC
100 atoms, 99 bonds, 1 residue, 1 model selected
> select #1:RCC,GPC
4100 atoms, 4059 bonds, 41 residues, 1 model selected
> close #1
> addh
Summary of feedback from adding hydrogens to
working_noh_real_space_refined.pdb #2.2
---
notes | No usable SEQRES records for working_noh_real_space_refined.pdb (#2.2)
chain AA; guessing termini instead
No usable SEQRES records for working_noh_real_space_refined.pdb (#2.2) chain
AB; guessing termini instead
No usable SEQRES records for working_noh_real_space_refined.pdb (#2.2) chain
AC; guessing termini instead
No usable SEQRES records for working_noh_real_space_refined.pdb (#2.2) chain
AD; guessing termini instead
No usable SEQRES records for working_noh_real_space_refined.pdb (#2.2) chain
AE; guessing termini instead
80 messages similar to the above omitted
Chain-initial residues that are actual N termini: /AA HIS 2, /AB HIS 2, /AC
HIS 2, /AD HIS 2, /AE HIS 2, /AF HIS 2, /AG HIS 2, /AH HIS 2, /AI HIS 2, /AJ
HIS 2, /AK HIS 2, /AL HIS 2, /AM HIS 2, /AN HIS 2, /AO HIS 2, /AP HIS 2, /AQ
HIS 2, /AR HIS 2, /AS HIS 2, /AT HIS 2, /AU HIS 2, /AV HIS 2, /AW HIS 2, /AX
HIS 2, /BA GLY 6, /BB GLY 5, /BC GLY 6, /BD GLY 5, /BE GLY 5, /BF GLY 6, /BG
GLY 6, /BH GLY 6, /BI GLY 5, /BJ GLY 6, /BK GLY 6, /BL GLY 6, /BM GLY 6, /BN
GLY 6, /BO GLY 6, /BP GLY 6, /BQ GLY 6, /BR GLY 6, /BS GLY 6, /BT GLY 6, /BU
GLY 6, /BV GLY 5, /BW GLY 5, /BX GLY 6, /C ALA 15, /H1 MET 1, /H2 SER 1, /L
ALA 1, /M MET 1, /aa HIS 2, /ab MET 1, /ac MET 1, /ad MET 1, /ae MET 1, /af
MET 1, /ag MET 1, /ah MET 1, /ai MET 1, /aj MET 1, /ak MET 1, /al MET 1, /am
MET 1, /an MET 1, /ao MET 1, /ap MET 1, /ba GLY 6, /bb GLY 6, /bc GLY 6, /bd
GLY 6, /be GLY 6, /bf GLY 6, /bg GLY 6, /bh GLY 6, /bi GLY 6, /bj GLY 6, /bk
GLY 6, /bl GLY 6, /bm GLY 6, /bn GLY 5, /bo GLY 6, /bp GLY 6
Chain-initial residues that are not actual N termini:
Chain-final residues that are actual C termini: /BA PHE 44, /BB PHE 44, /BC
PHE 44, /BD PHE 44, /BE PHE 44, /BF PHE 44, /BG PHE 44, /BH PHE 44, /BI PHE
44, /BJ PHE 44, /BK PHE 44, /BL PHE 44, /BM PHE 44, /BN PHE 44, /BO PHE 44,
/BP PHE 44, /BQ PHE 44, /BR PHE 44, /BS PHE 44, /BT PHE 44, /BU PHE 44, /BV
PHE 44, /BW PHE 44, /BX PHE 44, /C ARG 302, /H2 ILE 181, /L LYS 273, /ba PHE
44, /bb PHE 44, /bc PHE 44, /bd PHE 44, /be PHE 44, /bf PHE 44, /bg PHE 44,
/bh PHE 44, /bi PHE 44, /bj PHE 44, /bk PHE 44, /bl PHE 44, /bm PHE 44, /bn
PHE 44, /bo PHE 44, /bp PHE 44
Chain-final residues that are not actual C termini: /AA TYR 46, /AB TYR 46,
/AC TYR 46, /AD TYR 46, /AE TYR 46, /AF TYR 46, /AG TYR 46, /AH TYR 46, /AI
TYR 46, /AJ TYR 46, /AK TYR 46, /AL TYR 46, /AM TYR 46, /AN TYR 46, /AO TYR
46, /AP TYR 46, /AQ TYR 46, /AR TYR 46, /AS TYR 46, /AT TYR 46, /AU TYR 46,
/AV TYR 46, /AW TYR 46, /AX TYR 46, /H1 LYS 53, /M TYR 324, /aa ALA 60, /ab
ALA 60, /ac ALA 60, /ad ALA 60, /ae ALA 60, /af ALA 60, /ag ALA 60, /ah ALA
60, /ai ALA 60, /aj ALA 60, /ak ALA 60, /al ALA 60, /am ALA 60, /an ALA 60,
/ao ALA 60, /ap ALA 60
4864 hydrogen bonds
/AA TYR 46 is not terminus, removing H atom from 'C'
/AB TYR 46 is not terminus, removing H atom from 'C'
/AC TYR 46 is not terminus, removing H atom from 'C'
/AD TYR 46 is not terminus, removing H atom from 'C'
/AE TYR 46 is not terminus, removing H atom from 'C'
37 messages similar to the above omitted
43714 hydrogens added
> hide HC
> select ~protein
16594 atoms, 16919 bonds, 365 pseudobonds, 133 residues, 8 models selected
> select clear
> save old_model_in_map_1b.cxs
opened ChimeraX session
> isolde start
> set selectionWidth 4
Done loading forcefield
> select :GPC
4013 atoms, 3973 bonds, 40 residues, 1 model selected
> ui tool show Shell
/opt/UCSF/ChimeraX/lib/python3.7/site-packages/IPython/core/history.py:226:
UserWarning: IPython History requires SQLite, your history will not be saved
warn("IPython History requires SQLite, your history will not be saved")
> select clear
> delete sel
> delete sel
> delete sel
> delete sel
> save ready.cxs
> select gpc
Expected an objects specifier or a keyword
> select :GPC
4000 atoms, 3960 bonds, 40 residues, 1 model selected
> select :GPC
4000 atoms, 3960 bonds, 40 residues, 1 model selected
> show sel
> select clear
> show sel
> save ready.cxs
> volume gaussian #1 bfactor 50
> volume gaussian #2 bfactor 50
> clipper associate #1 toModel #2
> hide HC
> select #1
Nothing selected
> select #2
91607 atoms, 93634 bonds, 14 pseudobonds, 4857 residues, 16 models selected
> clipper set contourSensitivity 0.25
> select clear
> set bgColor white
> select #1
Nothing selected
> select #2
91607 atoms, 93634 bonds, 14 pseudobonds, 4857 residues, 16 models selected
> select clear
> select #2
91607 atoms, 93634 bonds, 14 pseudobonds, 4857 residues, 16 models selected
> select #1
Nothing selected
> select #2
91607 atoms, 93634 bonds, 14 pseudobonds, 4857 residues, 16 models selected
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
> select clear
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/mystery_helix/assigned_sequence.pdb
Summary of feedback from opening
/run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/mystery_helix/assigned_sequence.pdb
---
warnings | Start residue of secondary structure not found: HELIX 1 1 ILE B 5
TYR B 9 1 5
Start residue of secondary structure not found: HELIX 2 2 ARG B 22 ARG B 52 1
31
Start residue of secondary structure not found: HELIX 3 3 VAL B 55 ARG B 86 1
32
Chain information for assigned_sequence.pdb #1
---
Chain | Description
Ba | No description available
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/c_extra/sequence_assigned.pdb
Summary of feedback from opening
/run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/c_extra/sequence_assigned.pdb
---
warnings | Start residue of secondary structure not found: HELIX 1 1 THR C 19
GLY C 21 1 3
Start residue of secondary structure not found: HELIX 2 2 VAL C 28 VAL C 31 1
4
Start residue of secondary structure not found: HELIX 3 3 VAL C 32 HIS C 34 1
3
Start residue of secondary structure not found: HELIX 4 4 GLY C 61 ILE C 64 1
4
Start residue of secondary structure not found: HELIX 5 5 MET C 71 LYS C 74 1
4
3 messages similar to the above omitted
Chain information for sequence_assigned.pdb #3
---
Chain | Description
C1 | No description available
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/rc-
> dlh1-model1b_rearranged.pdb
Chain information for rc-dlh1-model1b_rearranged.pdb #4
---
Chain | Description
AA AB AC AD AE AF AG AH AI AJ AK AL AM AN AO AP AQ AR AS AT AU AV AW AX | No
description available
Aa Ab Ac Ad Ae Af Ah Ai Aj Ak Al Am An Ao Ap | No description available
Ag | No description available
B | No description available
BA BB BC BD BE BF BG BH BI BJ BK BL BM BN BO BP BQ BR BS BT BU BV BW BX Ba Bb
Bc Bd Be Bf Bg Bh Bi Bj Bk Bl Bm Bn Bo Bp | No description available
C | No description available
C1 | No description available
H1 | No description available
H2 | No description available
L | No description available
M | No description available
> hide #!4 models
> select #3
1472 atoms, 1487 bonds, 92 residues, 1 model selected
> save working.cxs
Taking snapshot of stepper: working_noh_real_space_refined.pdb
> hide #3 models
> select #1
1516 atoms, 1537 bonds, 99 residues, 1 model selected
> select #2/Ba
100 atoms, 99 bonds, 1 residue, 1 model selected
> view sel
> select #1
1516 atoms, 1537 bonds, 99 residues, 1 model selected
> select clear
> select /AA
1506 atoms, 1545 bonds, 4 pseudobonds, 96 residues, 3 models selected
> select /BA
1151 atoms, 1189 bonds, 4 pseudobonds, 79 residues, 3 models selected
> isolde stepto /AA
Multiple residues selected! Going to the first...
> select /AA
1506 atoms, 1545 bonds, 4 pseudobonds, 96 residues, 3 models selected
> select clear
> select clear
> select /AA
1521 atoms, 1560 bonds, 4 pseudobonds, 97 residues, 3 models selected
> select clear
> sequence chain /AA
Chains must have same sequence
> sequence chain #1/AA
Chains must have same sequence
> sequence chain #2/AA
Alignment identifier is 2.2/AA
> select /AA
1521 atoms, 1560 bonds, 4 pseudobonds, 97 residues, 3 models selected
> select clear
> select clear
> select /AA
1521 atoms, 1560 bonds, 4 pseudobonds, 97 residues, 3 models selected
> select clear
> select clear
> show #!4 models
> select #4/AS
540 atoms, 558 bonds, 4 pseudobonds, 49 residues, 2 models selected
> select clear
> select #4
51294 atoms, 53027 bonds, 354 pseudobonds, 5083 residues, 3 models selected
> style sel stick
Changed 51294 atom styles
> select clear
> hide #!4 models
> select clear
> isolde stepto /C
Multiple residues selected! Going to the first...
> select /C:15-200
4296 atoms, 4372 bonds, 372 residues, 2 models selected
> select clear
> show #!4 models
> hide #!4 models
> show #!4 models
> hide #!4 models
> isolde stepto
> isolde stepto
> isolde stepto
> show #!4 models
> hide #!4 models
reverting to start
> select /C1
4416 atoms, 4461 bonds, 276 residues, 3 models selected
> isolde ignore /C1:30-300
ISOLDE: currently ignoring 63 residues in model 2.2
ISOLDE: currently ignoring 63 residues in model 3
ISOLDE: currently ignoring 63 residues in model 4
> select /C:15-200
4296 atoms, 4372 bonds, 372 residues, 2 models selected
> select /M:311-400
359 atoms, 361 bonds, 28 residues, 2 models selected
> select clear
> isolde ~ignore
> show #!4 models
> hide #!4 models
> save working.cxs
Taking snapshot of stepper: working_noh_real_space_refined.pdb
> select /C:15-200
4296 atoms, 4372 bonds, 372 residues, 2 models selected
> select clear
> isolde stepto
> hide #1 models
> isolde ignore /C2
ISOLDE: currently ignoring 99 residues in model 2.2
> select /H2:120-129
241 atoms, 251 bonds, 20 residues, 2 models selected
> select clear
> select clear
> isolde ~ignore
> select up
22 atoms, 21 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 2.2
> isolde ~ignore
> select clear
> select up
24 atoms, 23 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 2.2
> isolde ~ignore
> select up
14 atoms, 13 bonds, 1 residue, 1 model selected
> select up
105 atoms, 108 bonds, 7 residues, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 8 residues in model 2.2
> select up
10 atoms, 9 bonds, 1 residue, 1 model selected
> select up
939 atoms, 951 bonds, 60 residues, 1 model selected
> select clear
> show #!4 models
> select clear
> hide #!4 models
> select clear
> select clear
> select clear
> select clear
> select clear
> save working.cxs
Taking snapshot of stepper: working_noh_real_space_refined.pdb
> select /C:15-200
4296 atoms, 4372 bonds, 372 residues, 2 models selected
> select clear
> isolde stepto /C
Multiple residues selected! Going to the first...
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> show #!4 models
> hide #!4 models
> isolde stepto
> isolde stepto
> isolde stepto
> show #!4 models
> hide #!4 models
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> show #!4 models
> hide #!4 models
> show #!4 models
> hide #!4 models
> select clear
> select up
14 atoms, 14 bonds, 1 residue, 1 model selected
> select up
450 atoms, 459 bonds, 30 residues, 1 model selected
> select clear
> select clear
> select /C:64-200
3217 atoms, 3269 bonds, 274 residues, 2 models selected
> select clear
> select clear
> select clear
> isolde stepto /C:64
Multiple residues selected! Going to the first...
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> delete sel
> select /C:99-200
2415 atoms, 2455 bonds, 204 residues, 2 models selected
> select clear
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> show #!4 models
> hide #!4 models
> show #!4 models
> hide #!4 models
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> show #!4 models
> hide #!4 models
> select /C:179-189
241 atoms, 241 bonds, 22 residues, 2 models selected
> select #2/C:179-189
162 atoms, 162 bonds, 11 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> save working.cxs
Taking snapshot of stepper: working_noh_real_space_refined.pdb
> select /C:200-400
2486 atoms, 2541 bonds, 217 residues, 2 models selected
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> show #!4 models
> save working.cxs
Taking snapshot of stepper: working_noh_real_space_refined.pdb
> select #4/C:303-313
83 atoms, 87 bonds, 11 residues, 1 model selected
> select clear
> hide #!4 models
> select #2/C:303-400
83 atoms, 87 bonds, 11 residues, 1 model selected
> show sel
> delete sell
Missing or invalid "atoms" argument: invalid atoms specifier
> delete sel
> addh #4
Summary of feedback from adding hydrogens to rc-dlh1-model1b_rearranged.pdb #4
---
warning | Skipped 8 atom(s) with bad connectivities; see log for details
notes | No usable SEQRES records for rc-dlh1-model1b_rearranged.pdb (#4) chain
AA; guessing termini instead
No usable SEQRES records for rc-dlh1-model1b_rearranged.pdb (#4) chain AB;
guessing termini instead
No usable SEQRES records for rc-dlh1-model1b_rearranged.pdb (#4) chain AC;
guessing termini instead
No usable SEQRES records for rc-dlh1-model1b_rearranged.pdb (#4) chain AD;
guessing termini instead
No usable SEQRES records for rc-dlh1-model1b_rearranged.pdb (#4) chain AE;
guessing termini instead
82 messages similar to the above omitted
Chain-initial residues that are actual N termini: rc-
dlh1-model1b_rearranged.pdb #4/AA HIS 2, rc-dlh1-model1b_rearranged.pdb #4/AB
HIS 2, rc-dlh1-model1b_rearranged.pdb #4/AC HIS 2, rc-
dlh1-model1b_rearranged.pdb #4/AD HIS 2, rc-dlh1-model1b_rearranged.pdb #4/AE
HIS 2, rc-dlh1-model1b_rearranged.pdb #4/AF HIS 2, rc-
dlh1-model1b_rearranged.pdb #4/AG HIS 2, rc-dlh1-model1b_rearranged.pdb #4/AH
HIS 2, rc-dlh1-model1b_rearranged.pdb #4/AI HIS 2, rc-
dlh1-model1b_rearranged.pdb #4/AJ HIS 2, rc-dlh1-model1b_rearranged.pdb #4/AK
HIS 2, rc-dlh1-model1b_rearranged.pdb #4/AL HIS 2, rc-
dlh1-model1b_rearranged.pdb #4/AM HIS 2, rc-dlh1-model1b_rearranged.pdb #4/AN
HIS 2, rc-dlh1-model1b_rearranged.pdb #4/AO HIS 2, rc-
dlh1-model1b_rearranged.pdb #4/AP HIS 2, rc-dlh1-model1b_rearranged.pdb #4/AQ
HIS 2, rc-dlh1-model1b_rearranged.pdb #4/AR HIS 2, rc-
dlh1-model1b_rearranged.pdb #4/AS HIS 2, rc-dlh1-model1b_rearranged.pdb #4/AT
HIS 2, rc-dlh1-model1b_rearranged.pdb #4/AU HIS 2, rc-
dlh1-model1b_rearranged.pdb #4/AV HIS 2, rc-dlh1-model1b_rearranged.pdb #4/AW
HIS 2, rc-dlh1-model1b_rearranged.pdb #4/AX HIS 2, rc-
dlh1-model1b_rearranged.pdb #4/Aa MET 1, rc-dlh1-model1b_rearranged.pdb #4/Ab
MET 1, rc-dlh1-model1b_rearranged.pdb #4/Ac MET 1, rc-
dlh1-model1b_rearranged.pdb #4/Ad MET 1, rc-dlh1-model1b_rearranged.pdb #4/Ae
MET 1, rc-dlh1-model1b_rearranged.pdb #4/Af MET 1, rc-
dlh1-model1b_rearranged.pdb #4/Ag MET 1, rc-dlh1-model1b_rearranged.pdb #4/Ah
MET 1, rc-dlh1-model1b_rearranged.pdb #4/Ai MET 1, rc-
dlh1-model1b_rearranged.pdb #4/Aj MET 1, rc-dlh1-model1b_rearranged.pdb #4/Ak
MET 1, rc-dlh1-model1b_rearranged.pdb #4/Al MET 1, rc-
dlh1-model1b_rearranged.pdb #4/Am MET 1, rc-dlh1-model1b_rearranged.pdb #4/An
MET 1, rc-dlh1-model1b_rearranged.pdb #4/Ao MET 1, rc-
dlh1-model1b_rearranged.pdb #4/Ap MET 1, rc-dlh1-model1b_rearranged.pdb #4/B
PRO 25, rc-dlh1-model1b_rearranged.pdb #4/BA MET 1, rc-
dlh1-model1b_rearranged.pdb #4/BB MET 1, rc-dlh1-model1b_rearranged.pdb #4/BC
MET 1, rc-dlh1-model1b_rearranged.pdb #4/BD MET 1, rc-
dlh1-model1b_rearranged.pdb #4/BE MET 1, rc-dlh1-model1b_rearranged.pdb #4/BF
MET 1, rc-dlh1-model1b_rearranged.pdb #4/BG MET 1, rc-
dlh1-model1b_rearranged.pdb #4/BH MET 1, rc-dlh1-model1b_rearranged.pdb #4/BI
MET 1, rc-dlh1-model1b_rearranged.pdb #4/BJ MET 1, rc-
dlh1-model1b_rearranged.pdb #4/BK MET 1, rc-dlh1-model1b_rearranged.pdb #4/BL
MET 1, rc-dlh1-model1b_rearranged.pdb #4/BM MET 1, rc-
dlh1-model1b_rearranged.pdb #4/BN MET 1, rc-dlh1-model1b_rearranged.pdb #4/BO
MET 1, rc-dlh1-model1b_rearranged.pdb #4/BP MET 1, rc-
dlh1-model1b_rearranged.pdb #4/BQ MET 1, rc-dlh1-model1b_rearranged.pdb #4/BR
MET 1, rc-dlh1-model1b_rearranged.pdb #4/BS MET 1, rc-
dlh1-model1b_rearranged.pdb #4/BT MET 1, rc-dlh1-model1b_rearranged.pdb #4/BU
MET 1, rc-dlh1-model1b_rearranged.pdb #4/BV MET 1, rc-
dlh1-model1b_rearranged.pdb #4/BW MET 1, rc-dlh1-model1b_rearranged.pdb #4/BX
MET 1, rc-dlh1-model1b_rearranged.pdb #4/Ba MET 1, rc-
dlh1-model1b_rearranged.pdb #4/Bb MET 1, rc-dlh1-model1b_rearranged.pdb #4/Bc
MET 1, rc-dlh1-model1b_rearranged.pdb #4/Bd MET 1, rc-
dlh1-model1b_rearranged.pdb #4/Be MET 1, rc-dlh1-model1b_rearranged.pdb #4/Bf
MET 1, rc-dlh1-model1b_rearranged.pdb #4/Bg MET 1, rc-
dlh1-model1b_rearranged.pdb #4/Bh MET 1, rc-dlh1-model1b_rearranged.pdb #4/Bi
MET 1, rc-dlh1-model1b_rearranged.pdb #4/Bj MET 1, rc-
dlh1-model1b_rearranged.pdb #4/Bk MET 1, rc-dlh1-model1b_rearranged.pdb #4/Bl
MET 1, rc-dlh1-model1b_rearranged.pdb #4/Bm MET 1, rc-
dlh1-model1b_rearranged.pdb #4/Bn MET 1, rc-dlh1-model1b_rearranged.pdb #4/Bo
MET 1, rc-dlh1-model1b_rearranged.pdb #4/Bp MET 1, rc-
dlh1-model1b_rearranged.pdb #4/C ALA 15, rc-dlh1-model1b_rearranged.pdb #4/H1
MET 1, rc-dlh1-model1b_rearranged.pdb #4/H2 SER 1, rc-
dlh1-model1b_rearranged.pdb #4/L ALA 1, rc-dlh1-model1b_rearranged.pdb #4/M
MET 1
Chain-initial residues that are not actual N termini: rc-
dlh1-model1b_rearranged.pdb #4/C1 GLN 1, rc-dlh1-model1b_rearranged.pdb #4/H2
ARG 38
Chain-final residues that are actual C termini: rc-dlh1-model1b_rearranged.pdb
#4/BA PHE 38, rc-dlh1-model1b_rearranged.pdb #4/BB PHE 38, rc-
dlh1-model1b_rearranged.pdb #4/BC PHE 38, rc-dlh1-model1b_rearranged.pdb #4/BD
PHE 38, rc-dlh1-model1b_rearranged.pdb #4/BE PHE 38, rc-
dlh1-model1b_rearranged.pdb #4/BF PHE 38, rc-dlh1-model1b_rearranged.pdb #4/BG
PHE 38, rc-dlh1-model1b_rearranged.pdb #4/BH PHE 38, rc-
dlh1-model1b_rearranged.pdb #4/BI PHE 38, rc-dlh1-model1b_rearranged.pdb #4/BJ
PHE 38, rc-dlh1-model1b_rearranged.pdb #4/BK PHE 38, rc-
dlh1-model1b_rearranged.pdb #4/BL PHE 38, rc-dlh1-model1b_rearranged.pdb #4/BM
PHE 38, rc-dlh1-model1b_rearranged.pdb #4/BN PHE 38, rc-
dlh1-model1b_rearranged.pdb #4/BO PHE 38, rc-dlh1-model1b_rearranged.pdb #4/BP
PHE 38, rc-dlh1-model1b_rearranged.pdb #4/BQ PHE 38, rc-
dlh1-model1b_rearranged.pdb #4/BR PHE 38, rc-dlh1-model1b_rearranged.pdb #4/BS
PHE 38, rc-dlh1-model1b_rearranged.pdb #4/BT PHE 38, rc-
dlh1-model1b_rearranged.pdb #4/BU PHE 38, rc-dlh1-model1b_rearranged.pdb #4/BV
PHE 38, rc-dlh1-model1b_rearranged.pdb #4/BW PHE 38, rc-
dlh1-model1b_rearranged.pdb #4/BX PHE 38, rc-dlh1-model1b_rearranged.pdb #4/Ba
PHE 38, rc-dlh1-model1b_rearranged.pdb #4/Bb PHE 38, rc-
dlh1-model1b_rearranged.pdb #4/Bc PHE 38, rc-dlh1-model1b_rearranged.pdb #4/Bd
PHE 38, rc-dlh1-model1b_rearranged.pdb #4/Be PHE 38, rc-
dlh1-model1b_rearranged.pdb #4/Bf PHE 38, rc-dlh1-model1b_rearranged.pdb #4/Bg
PHE 38, rc-dlh1-model1b_rearranged.pdb #4/Bh PHE 38, rc-
dlh1-model1b_rearranged.pdb #4/Bi PHE 38, rc-dlh1-model1b_rearranged.pdb #4/Bj
PHE 38, rc-dlh1-model1b_rearranged.pdb #4/Bk PHE 38, rc-
dlh1-model1b_rearranged.pdb #4/Bl PHE 38, rc-dlh1-model1b_rearranged.pdb #4/Bm
PHE 38, rc-dlh1-model1b_rearranged.pdb #4/Bn PHE 38, rc-
dlh1-model1b_rearranged.pdb #4/Bo PHE 38, rc-dlh1-model1b_rearranged.pdb #4/Bp
PHE 38
Chain-final residues that are not actual C termini: rc-
dlh1-model1b_rearranged.pdb #4/AA PRO 48, rc-dlh1-model1b_rearranged.pdb #4/AB
PRO 48, rc-dlh1-model1b_rearranged.pdb #4/AC PRO 48, rc-
dlh1-model1b_rearranged.pdb #4/AD PRO 48, rc-dlh1-model1b_rearranged.pdb #4/AE
PRO 48, rc-dlh1-model1b_rearranged.pdb #4/AF PRO 48, rc-
dlh1-model1b_rearranged.pdb #4/AG PRO 48, rc-dlh1-model1b_rearranged.pdb #4/AH
PRO 48, rc-dlh1-model1b_rearranged.pdb #4/AI PRO 48, rc-
dlh1-model1b_rearranged.pdb #4/AJ PRO 48, rc-dlh1-model1b_rearranged.pdb #4/AK
PRO 48, rc-dlh1-model1b_rearranged.pdb #4/AL PRO 48, rc-
dlh1-model1b_rearranged.pdb #4/AM PRO 48, rc-dlh1-model1b_rearranged.pdb #4/AN
PRO 48, rc-dlh1-model1b_rearranged.pdb #4/AO PRO 48, rc-
dlh1-model1b_rearranged.pdb #4/AP PRO 48, rc-dlh1-model1b_rearranged.pdb #4/AQ
PRO 48, rc-dlh1-model1b_rearranged.pdb #4/AR PRO 48, rc-
dlh1-model1b_rearranged.pdb #4/AS PRO 48, rc-dlh1-model1b_rearranged.pdb #4/AT
PRO 48, rc-dlh1-model1b_rearranged.pdb #4/AU PRO 48, rc-
dlh1-model1b_rearranged.pdb #4/AV PRO 48, rc-dlh1-model1b_rearranged.pdb #4/AW
PRO 48, rc-dlh1-model1b_rearranged.pdb #4/AX PRO 48, rc-
dlh1-model1b_rearranged.pdb #4/Aa ALA 60, rc-dlh1-model1b_rearranged.pdb #4/Ab
ALA 60, rc-dlh1-model1b_rearranged.pdb #4/Ac ALA 60, rc-
dlh1-model1b_rearranged.pdb #4/Ad ALA 60, rc-dlh1-model1b_rearranged.pdb #4/Ae
ALA 60, rc-dlh1-model1b_rearranged.pdb #4/Af ALA 60, rc-
dlh1-model1b_rearranged.pdb #4/Ag LEU 67, rc-dlh1-model1b_rearranged.pdb #4/Ah
ALA 60, rc-dlh1-model1b_rearranged.pdb #4/Ai ALA 60, rc-
dlh1-model1b_rearranged.pdb #4/Aj ALA 60, rc-dlh1-model1b_rearranged.pdb #4/Ak
ALA 60, rc-dlh1-model1b_rearranged.pdb #4/Al ALA 60, rc-
dlh1-model1b_rearranged.pdb #4/Am ALA 60, rc-dlh1-model1b_rearranged.pdb #4/An
ALA 60, rc-dlh1-model1b_rearranged.pdb #4/Ao ALA 60, rc-
dlh1-model1b_rearranged.pdb #4/Ap ALA 60, rc-dlh1-model1b_rearranged.pdb #4/B
ILE 123, rc-dlh1-model1b_rearranged.pdb #4/C ILE 313, rc-
dlh1-model1b_rearranged.pdb #4/C1 LEU 92, rc-dlh1-model1b_rearranged.pdb #4/H1
HIS 67, rc-dlh1-model1b_rearranged.pdb #4/H2 ILE 181, rc-
dlh1-model1b_rearranged.pdb #4/H2 ARG 36, rc-dlh1-model1b_rearranged.pdb #4/L
TRP 272, rc-dlh1-model1b_rearranged.pdb #4/M TYR 324
Skipping possible acceptor with bad geometry: rc-dlh1-model1b_rearranged.pdb
#4/C ARG 302 O
Wrong number of grandchild atoms for phi/psi acceptor rc-
dlh1-model1b_rearranged.pdb #4/C ARG 302 O
Skipping possible acceptor with bad geometry: rc-dlh1-model1b_rearranged.pdb
#4/C ARG 302 OXT
Wrong number of grandchild atoms for phi/psi acceptor rc-
dlh1-model1b_rearranged.pdb #4/C ARG 302 OXT
Skipping possible acceptor with bad geometry: rc-dlh1-model1b_rearranged.pdb
#4/C ARG 302 OXT
Wrong number of grandchild atoms for phi/psi acceptor rc-
dlh1-model1b_rearranged.pdb #4/C ARG 302 OXT
Skipping possible acceptor with bad geometry: rc-dlh1-model1b_rearranged.pdb
#4/C ARG 302 O
Wrong number of grandchild atoms for phi/psi acceptor rc-
dlh1-model1b_rearranged.pdb #4/C ARG 302 O
Skipping possible acceptor with bad geometry: rc-dlh1-model1b_rearranged.pdb
#4/C ARG 302 O
Wrong number of grandchild atoms for phi/psi acceptor rc-
dlh1-model1b_rearranged.pdb #4/C ARG 302 O
3 messages similar to the above omitted
4768 hydrogen bonds
Adding 'H' to rc-dlh1-model1b_rearranged.pdb #4/H2 ARG 38
rc-dlh1-model1b_rearranged.pdb #4/AA PRO 48 is not terminus, removing H atom
from 'C'
rc-dlh1-model1b_rearranged.pdb #4/AB PRO 48 is not terminus, removing H atom
from 'C'
rc-dlh1-model1b_rearranged.pdb #4/AC PRO 48 is not terminus, removing H atom
from 'C'
rc-dlh1-model1b_rearranged.pdb #4/AD PRO 48 is not terminus, removing H atom
from 'C'
rc-dlh1-model1b_rearranged.pdb #4/AE PRO 48 is not terminus, removing H atom
from 'C'
42 messages similar to the above omitted
44090 hydrogens added
> select #2/C:303-400
157 atoms, 161 bonds, 11 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> show #!4 models
> hide #!4 models
> select up
12 atoms, 11 bonds, 1 residue, 1 model selected
> select up
874 atoms, 890 bonds, 53 residues, 1 model selected
> select #2/H1:3-100
838 atoms, 854 bonds, 51 residues, 1 model selected
> show #!4 models
> hide #!4 models
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> show #!4 models
> hide #!4 models
> select up
38 atoms, 40 bonds, 2 residues, 1 model selected
> select clear
> select clear
> select #1
1516 atoms, 1537 bonds, 99 residues, 1 model selected
> select #2
94789 atoms, 96857 bonds, 14 pseudobonds, 5061 residues, 22 models selected
> style sel sphere
Changed 94789 atom styles
> select clear
> style #!2.2 stick
Changed 94789 atom styles
> delete sel
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> delete sel
> delete sel
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> delete sel
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select #2/H2
2777 atoms, 2816 bonds, 180 residues, 1 model selected
> select clear
> isolde jumpto
> select #2/H2:3-100
1476 atoms, 1499 bonds, 97 residues, 1 model selected
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select #2/H2:3-100
1476 atoms, 1499 bonds, 97 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select #2/H1:95-205
Nothing selected
> select #2/H1:95-205
Nothing selected
> select #2/H2:95-205
1376 atoms, 1394 bonds, 87 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> save working.cxs
Taking snapshot of stepper: working_noh_real_space_refined.pdb
> isolde stepto
> select #2/L:1-100
1563 atoms, 1592 bonds, 100 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select #2/L:100-300
2756 atoms, 2816 bonds, 174 residues, 1 model selected
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> swapaa mousemode sel ILE
> select up
19 atoms, 18 bonds, 1 residue, 1 model selected
> color sel bychain
> color sel byhetero
> select #2/L:227-400
777 atoms, 798 bonds, 47 residues, 1 model selected
> hide HC
> select up
15 atoms, 14 bonds, 1 residue, 1 model selected
> color sel byhetero
> select clear
> color bychain
> color byhetero
> hide HC
> select #2/L:227-400
777 atoms, 798 bonds, 47 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> show H&~HC
> select clear
> show H&~HC
> show ~H
> select #2/L:227-400
777 atoms, 798 bonds, 47 residues, 1 model selected
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select #2/M
5833 atoms, 5950 bonds, 6 pseudobonds, 330 residues, 2 models selected
> select #2/M:1-50
763 atoms, 772 bonds, 50 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select #2/M:58-158
1664 atoms, 1698 bonds, 101 residues, 1 model selected
> select clear
> select #2/M:58-158
1664 atoms, 1698 bonds, 101 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> sequence chain #2/M
Alignment identifier is 2.2/M
> select #2/M:58-158
1665 atoms, 1699 bonds, 101 residues, 1 model selected
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> delete sel
> select #2/M:178-400
2337 atoms, 2378 bonds, 147 residues, 1 model selected
> select clear
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> save working.cxs
Taking snapshot of stepper: working_noh_real_space_refined.pdb
> show #!4 models
> hide #!4 models
> swapaa mousemode sel GLY
> select up
16 atoms, 15 bonds, 1 residue, 1 model selected
> select up
255 atoms, 259 bonds, 18 residues, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 26 residues in model 2.2
> select clear
> isolde ~ignore
> select up
23 atoms, 21 bonds, 2 residues, 1 model selected
> select clear
> delete sel
> delete sel
> select clear
> save working.cxs
Taking snapshot of stepper: working_noh_real_space_refined.pdb
> ui tool show "Build Structure"
> build start peptide "custom built" FPSYVVPQNATMPDTAAAPIVTDSITTDSTKTGGTQ
> -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0
> -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0
> -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0
> -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0
> -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0
> -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0
> rotLib Dunbrack
Chain information for custom built #5
---
Chain | Description
A | No description available
> build start peptide "custom built" FPSYVVPQNATMPDTAAAPIVTDSITTDSTKTGGTQ
> -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0
> -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0
> -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0
> -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0
> -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0
> -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0 -57.0,-47.0
> rotLib Dunbrack
Chain information for custom built #6
---
Chain | Description
A | No description available
> close #5
> select #6
258 atoms, 263 bonds, 36 residues, 1 model selected
> show sel&:1
> ui mousemode right "translate selected models"
> ui mousemode right "rotate selected models"
> ui mousemode right "translate selected models"
> ui mousemode right "rotate selected models"
> ui mousemode right "translate selected models"
> hide #6 models
> delete #2/M:332-367
> addh #6
Summary of feedback from adding hydrogens to custom built #6
---
notes | No usable SEQRES records for custom built (#6) chain A; guessing
termini instead
Chain-initial residues that are actual N termini: custom built #6/A PHE 1
Chain-initial residues that are not actual N termini:
Chain-final residues that are actual C termini: custom built #6/A GLN 36
Chain-final residues that are not actual C termini:
37 hydrogen bonds
250 hydrogens added
> isolde ignore #2/C
ISOLDE: currently ignoring 303 residues in model 2.2
> select #2/M:332-367
506 atoms, 511 bonds, 36 residues, 1 model selected
> hide HC
> delete #2/M:339-367
> isolde ~ignore
> select clear
> select clear
> select up
11 atoms, 10 bonds, 1 residue, 1 model selected
> select up
92 atoms, 94 bonds, 6 residues, 1 model selected
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select up
14 atoms, 14 bonds, 1 residue, 1 model selected
> delete sel
> save ser_M331_O_glycan.jpg
> open 3e80
3e80 title:
Structure of Heparinase II complexed with heparan sulfate degradation
disaccharide product [more info...]
Chain information for 3e80 #5
---
Chain | Description
A B C | Heparinase II protein
Non-standard residues in 3e80 #5
---
GCD — 4-deoxy-alpha-L-threo-hex-4-enopyranuronic acid
GCU — alpha-D-glucopyranuronic acid
MAN — alpha-D-mannopyranose
NDG — 2-acetamido-2-deoxy-alpha-D-glucopyranose
PO4 — phosphate ion
RAM — alpha-L-rhamnopyranose
XYS — alpha-D-xylopyranose
ZN — zinc ion
3e80 mmCIF Assemblies
---
1| author_and_software_defined_assembly
2| author_and_software_defined_assembly
3| author_and_software_defined_assembly
> addh #5
Summary of feedback from adding hydrogens to 3e80 #5
---
warning | Not adding hydrogens to 3e80 #5/C LEU 732 CB because it is missing
heavy-atom bond partners
notes | Termini for 3e80 (#5) chain A determined from SEQRES records
Termini for 3e80 (#5) chain B determined from SEQRES records
Termini for 3e80 (#5) chain C determined from SEQRES records
Chain-initial residues that are actual N termini:
Chain-initial residues that are not actual N termini: 3e80 #5/A PCA 26, 3e80
#5/B PCA 26, 3e80 #5/C PCA 26
Chain-final residues that are actual C termini: 3e80 #5/A ARG 772, 3e80 #5/B
ARG 772, 3e80 #5/C ARG 772
Chain-final residues that are not actual C termini:
Missing OXT added to C-terminal residue 3e80 #5/A ARG 772
Missing OXT added to C-terminal residue 3e80 #5/B ARG 772
Missing OXT added to C-terminal residue 3e80 #5/C ARG 772
3540 hydrogen bonds
Adding 'H' to 3e80 #5/A PCA 26
Adding 'H' to 3e80 #5/B PCA 26
Adding 'H' to 3e80 #5/C PCA 26
18911 hydrogens added
> select #5
37630 atoms, 37586 bonds, 17 pseudobonds, 2807 residues, 2 models selected
> hide #!2 models
> select clear
> select clear
> select #5/B:134|(#5/E:1-4)
90 atoms, 93 bonds, 5 residues, 1 model selected
> hide #5&~sel
> ~cartoon #5
> show #!2 models
> ui mousemode right "translate selected models"
> ui mousemode right "rotate selected models"
> ui mousemode right "translate selected models"
> ui mousemode right "rotate selected models"
> ui mousemode right "translate selected models"
> select (#5/E:1-4)
77 atoms, 80 bonds, 4 residues, 1 model selected
> save working.cxs
Taking snapshot of stepper: working_noh_real_space_refined.pdb
Restoring stepper: working_noh_real_space_refined.pdb
opened ChimeraX session
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1b/working.pdb
Summary of feedback from opening
/run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1b/working.pdb
---
warnings | End residue of secondary structure not found: HELIX 55 55 THR C 19
GLY C 21 1 3
Start residue of secondary structure not found: HELIX 56 56 VAL C 28 HIS C 34
1 7
Start residue of secondary structure not found: HELIX 57 57 ILE C 53 ASN C 55
1 3
Start residue of secondary structure not found: HELIX 58 58 ILE C 64 TRP C 69
1 6
Start residue of secondary structure not found: HELIX 59 59 ARG C 75 ARG C 81
1 7
Start residue of secondary structure not found: HELIX 60 60 ALA C 89 GLN C 91
1 3
5 messages similar to the above omitted
Cannot find LINK/SSBOND residue GCU (2 )
Cannot find LINK/SSBOND residue XYS (3 )
Cannot find LINK/SSBOND residue RAM (4 )
Cannot find LINK/SSBOND residue MAN (1 )
Cannot find LINK/SSBOND residue GCU (2 )
3 messages similar to the above omitted
Chain information for working.pdb #7
---
Chain | Description
AA | No description available
AB AC AD AE AF AG AH AI AJ AK AL AM AN AO AP AQ AR AS AT AU AV AW AX | No
description available
BA BC BF BG BH BJ BK BL BM BN BO BP BQ BR BS BT BU BX ba bb bc bd be bf bg bh
bi bj bk bl bm bo bp | No description available
BB BD BE BI BV BW bn | No description available
C | No description available
C1 | No description available
C2 | No description available
H1 | No description available
H2 | No description available
L | No description available
M | No description available
aa | No description available
ab ac ad ae af ag ah ai aj am an ao ap | No description available
ak | No description available
> select clear
> hide #7&protein
> hide #!5 models
> select #7/CG
78 atoms, 81 bonds, 4 residues, 1 model selected
> hide #!7 models
> delete sel
> select clear
> select clear
> view #2/M:330
> show #!7 models
> select #7/MG
78 atoms, 81 bonds, 4 residues, 1 model selected
> hide #!7 models
> delete sel
> select clear
> select clear
> save M331_glycan.jpg
> save working.cxs
Taking snapshot of stepper: working_noh_real_space_refined.pdb
> select #2/ak
1079 atoms, 1099 bonds, 61 residues, 1 model selected
> ui tool show "Build Structure"
> build start peptide "custom built" AAEMSPLPPGR -139.0,135.0 -139.0,135.0
> -139.0,135.0 -139.0,135.0 -139.0,135.0 -139.0,135.0 -139.0,135.0
> -139.0,135.0 -139.0,135.0 -139.0,135.0 -139.0,135.0 rotLib Dunbrack
Chain information for custom built #8
---
Chain | Description
A | No description available
> addh #8
Summary of feedback from adding hydrogens to custom built #8
---
notes | No usable SEQRES records for custom built (#8) chain A; guessing
termini instead
Chain-initial residues that are actual N termini: custom built #8/A ALA 1
Chain-initial residues that are not actual N termini:
Chain-final residues that are actual C termini: custom built #8/A ARG 11
Chain-final residues that are not actual C termini:
0 hydrogen bonds
80 hydrogens added
> select #8
158 atoms, 160 bonds, 11 residues, 1 model selected
> show sel
> ui mousemode right "translate selected atoms"
> ui mousemode right "rotate selected models"
> ui mousemode right "translate selected models"
> ui mousemode right "rotate selected models"
> ui mousemode right "translate selected models"
> ui mousemode right "rotate selected models"
> ui mousemode right "translate selected models"
> save working_2.cxs
Taking snapshot of stepper: working_noh_real_space_refined.pdb
Restoring stepper: working_noh_real_space_refined.pdb
opened ChimeraX session
> save
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1b/working_2.pdb
> models #2
> close #2
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1b/working_2.pdb
Summary of feedback from opening
/run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1b/working_2.pdb
---
warnings | End residue of secondary structure not found: HELIX 55 55 THR C 19
GLY C 21 1 3
Start residue of secondary structure not found: HELIX 56 56 VAL C 28 HIS C 34
1 7
Start residue of secondary structure not found: HELIX 57 57 ILE C 53 ASN C 55
1 3
Start residue of secondary structure not found: HELIX 58 58 ILE C 64 TRP C 69
1 6
Start residue of secondary structure not found: HELIX 59 59 ARG C 75 ARG C 81
1 7
Start residue of secondary structure not found: HELIX 60 60 ALA C 89 GLN C 91
1 3
5 messages similar to the above omitted
Cannot find LINK/SSBOND residue GCU (2 )
Cannot find LINK/SSBOND residue XYS (3 )
Cannot find LINK/SSBOND residue RAM (4 )
Cannot find LINK/SSBOND residue MAN (1 )
Cannot find LINK/SSBOND residue GCU (2 )
3 messages similar to the above omitted
Chain information for working_2.pdb #8
---
Chain | Description
AA | No description available
AB AC AD AE AF AG AH AI AJ AK AL AM AN AO AP AQ AR AS AT AU AV AW AX | No
description available
BA BC BF BG BH BJ BK BL BM BN BO BP BQ BR BS BT BU BX ba bb bc bd be bf bg bh
bi bj bk bl bm bo bp | No description available
BB BD BE BI BV BW bn | No description available
C | No description available
C1 | No description available
C2 | No description available
H1 | No description available
H2 | No description available
L | No description available
M | No description available
aa | No description available
ab ac ad ae af ag ah ai aj ak al am an ao ap | No description available
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/class1b_26A.mrc
Opened class1b_26A.mrc, grid size 400,400,400, pixel 0.999, shown at level
0.0224, step 2, values float32
> clipper associate #9 toModel #8
Chain information for working_2.pdb
---
Chain | Description
8.2/AA | No description available
8.2/AB 8.2/AC 8.2/AD 8.2/AE 8.2/AF 8.2/AG 8.2/AH 8.2/AI 8.2/AJ 8.2/AK 8.2/AL
8.2/AM 8.2/AN 8.2/AO 8.2/AP 8.2/AQ 8.2/AR 8.2/AS 8.2/AT 8.2/AU 8.2/AV 8.2/AW
8.2/AX | No description available
8.2/BA 8.2/BC 8.2/BF 8.2/BG 8.2/BH 8.2/BJ 8.2/BK 8.2/BL 8.2/BM 8.2/BN 8.2/BO
8.2/BP 8.2/BQ 8.2/BR 8.2/BS 8.2/BT 8.2/BU 8.2/BX 8.2/ba 8.2/bb 8.2/bc 8.2/bd
8.2/be 8.2/bf 8.2/bg 8.2/bh 8.2/bi 8.2/bj 8.2/bk 8.2/bl 8.2/bm 8.2/bo 8.2/bp |
No description available
8.2/BB 8.2/BD 8.2/BE 8.2/BI 8.2/BV 8.2/BW 8.2/bn | No description available
8.2/C | No description available
8.2/C1 | No description available
8.2/C2 | No description available
8.2/H1 | No description available
8.2/H2 | No description available
8.2/L | No description available
8.2/M | No description available
8.2/aa | No description available
8.2/ab 8.2/ac 8.2/ad 8.2/ae 8.2/af 8.2/ag 8.2/ah 8.2/ai 8.2/aj 8.2/ak 8.2/al
8.2/am 8.2/an 8.2/ao 8.2/ap | No description available
> close #1
> close #2
> select #3
508 atoms, 513 bonds, 36 residues, 8 models selected
> hide #!4 models
> select #8/ak
1079 atoms, 1095 bonds, 4 pseudobonds, 61 residues, 2 models selected
> hide #!3 models
> show #!3 models
> close #4
> view #3
> show #3
> hide #!3 models
> show #!3 models
> view #3
> close #3
> close #5
> close #6
> close #7
> volume gaussian #8 bfactor 50
> clipper associate #1 toModel #8
> select clear
> save reopened.cxs
Taking snapshot of stepper: working_noh_real_space_refined.pdb
Traceback (most recent call last):
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 280, in process
data = sm.take_snapshot(obj, session, self.state_flags)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/atomic/molobject.py", line 633, in take_snapshot
data = {'structure': self.structure,
File "cymol.pyx", line 1437, in
chimerax.atomic.cymol.CyResidue.structure.__get__
RuntimeError: Residue already deleted
The above exception was the direct cause of the following exception:
Traceback (most recent call last):
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 259, in discovery
self.processed[key] = self.process(obj, parents)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 283, in process
raise RuntimeError(msg) from e
RuntimeError: Error while saving session data for 'isolde residue stepper 1'
-> <chimerax.isolde.navigate.ResidueStepper object at 0x7f42c411fd50> ->
<chimerax.atomic.molobject.Residue object at 0x7f4301f68aa0>
During handling of the above exception, another exception occurred:
Traceback (most recent call last):
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 826, in save
session.save(output, version=version, include_maps=include_maps)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 583, in save
mgr.discovery(self._state_containers)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 261, in discovery
raise ValueError("error processing: %s: %s" % (_obj_stack(parents, obj), e))
ValueError: error processing: 'isolde residue stepper 1' ->
<chimerax.isolde.navigate.ResidueStepper object at 0x7f42c411fd50> ->
<chimerax.atomic.molobject.Residue object at 0x7f4301f68aa0>: Error while
saving session data for 'isolde residue stepper 1' ->
<chimerax.isolde.navigate.ResidueStepper object at 0x7f42c411fd50> ->
<chimerax.atomic.molobject.Residue object at 0x7f4301f68aa0>
ValueError: error processing: 'isolde residue stepper 1' -> -> : Error while
saving session data for 'isolde residue stepper 1' -> ->
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 261, in discovery
raise ValueError("error processing: %s: %s" % (_obj_stack(parents, obj), e))
See log for complete Python traceback.
Traceback (most recent call last):
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 280, in process
data = sm.take_snapshot(obj, session, self.state_flags)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/atomic/molobject.py", line 633, in take_snapshot
data = {'structure': self.structure,
File "cymol.pyx", line 1437, in
chimerax.atomic.cymol.CyResidue.structure.__get__
RuntimeError: Residue already deleted
The above exception was the direct cause of the following exception:
Traceback (most recent call last):
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 259, in discovery
self.processed[key] = self.process(obj, parents)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 283, in process
raise RuntimeError(msg) from e
RuntimeError: Error while saving session data for 'isolde residue stepper 1'
-> <chimerax.isolde.navigate.ResidueStepper object at 0x7f42c411fd50> ->
<chimerax.atomic.molobject.Residue object at 0x7f4301f68aa0>
During handling of the above exception, another exception occurred:
Traceback (most recent call last):
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/cmd_line/tool.py", line 275, in execute
cmd.run(cmd_text)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/commands/cli.py", line 2805, in run
result = ci.function(session, **kw_args)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/save_command/cmd.py", line 66, in cmd_save
Command(session, registry=registry).run(provider_cmd_text, log=log)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/commands/cli.py", line 2805, in run
result = ci.function(session, **kw_args)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/save_command/cmd.py", line 79, in provider_save
mgr).save(session, path, **provider_kw)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core_formats/__init__.py", line 79, in save
return cxs_save(session, path, **kw)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 826, in save
session.save(output, version=version, include_maps=include_maps)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 583, in save
mgr.discovery(self._state_containers)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 261, in discovery
raise ValueError("error processing: %s: %s" % (_obj_stack(parents, obj), e))
ValueError: error processing: 'isolde residue stepper 1' ->
<chimerax.isolde.navigate.ResidueStepper object at 0x7f42c411fd50> ->
<chimerax.atomic.molobject.Residue object at 0x7f4301f68aa0>: Error while
saving session data for 'isolde residue stepper 1' ->
<chimerax.isolde.navigate.ResidueStepper object at 0x7f42c411fd50> ->
<chimerax.atomic.molobject.Residue object at 0x7f4301f68aa0>
ValueError: error processing: 'isolde residue stepper 1' -> -> : Error while
saving session data for 'isolde residue stepper 1' -> ->
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 261, in discovery
raise ValueError("error processing: %s: %s" % (_obj_stack(parents, obj), e))
See log for complete Python traceback.
> isolde stepto sel
> select up
10 atoms, 9 bonds, 1 residue, 1 model selected
> save reopened.cxs
Taking snapshot of stepper: working_2.pdb
Taking snapshot of stepper: working_noh_real_space_refined.pdb
Traceback (most recent call last):
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 280, in process
data = sm.take_snapshot(obj, session, self.state_flags)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/atomic/molobject.py", line 633, in take_snapshot
data = {'structure': self.structure,
File "cymol.pyx", line 1437, in
chimerax.atomic.cymol.CyResidue.structure.__get__
RuntimeError: Residue already deleted
The above exception was the direct cause of the following exception:
Traceback (most recent call last):
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 259, in discovery
self.processed[key] = self.process(obj, parents)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 283, in process
raise RuntimeError(msg) from e
RuntimeError: Error while saving session data for 'isolde residue stepper 1'
-> <chimerax.isolde.navigate.ResidueStepper object at 0x7f42c411fd50> ->
<chimerax.atomic.molobject.Residue object at 0x7f4301f68aa0>
During handling of the above exception, another exception occurred:
Traceback (most recent call last):
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 826, in save
session.save(output, version=version, include_maps=include_maps)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 583, in save
mgr.discovery(self._state_containers)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 261, in discovery
raise ValueError("error processing: %s: %s" % (_obj_stack(parents, obj), e))
ValueError: error processing: 'isolde residue stepper 1' ->
<chimerax.isolde.navigate.ResidueStepper object at 0x7f42c411fd50> ->
<chimerax.atomic.molobject.Residue object at 0x7f4301f68aa0>: Error while
saving session data for 'isolde residue stepper 1' ->
<chimerax.isolde.navigate.ResidueStepper object at 0x7f42c411fd50> ->
<chimerax.atomic.molobject.Residue object at 0x7f4301f68aa0>
ValueError: error processing: 'isolde residue stepper 1' -> -> : Error while
saving session data for 'isolde residue stepper 1' -> ->
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 261, in discovery
raise ValueError("error processing: %s: %s" % (_obj_stack(parents, obj), e))
See log for complete Python traceback.
Traceback (most recent call last):
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 280, in process
data = sm.take_snapshot(obj, session, self.state_flags)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/atomic/molobject.py", line 633, in take_snapshot
data = {'structure': self.structure,
File "cymol.pyx", line 1437, in
chimerax.atomic.cymol.CyResidue.structure.__get__
RuntimeError: Residue already deleted
The above exception was the direct cause of the following exception:
Traceback (most recent call last):
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 259, in discovery
self.processed[key] = self.process(obj, parents)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 283, in process
raise RuntimeError(msg) from e
RuntimeError: Error while saving session data for 'isolde residue stepper 1'
-> <chimerax.isolde.navigate.ResidueStepper object at 0x7f42c411fd50> ->
<chimerax.atomic.molobject.Residue object at 0x7f4301f68aa0>
During handling of the above exception, another exception occurred:
Traceback (most recent call last):
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/cmd_line/tool.py", line 275, in execute
cmd.run(cmd_text)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/commands/cli.py", line 2805, in run
result = ci.function(session, **kw_args)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/save_command/cmd.py", line 66, in cmd_save
Command(session, registry=registry).run(provider_cmd_text, log=log)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/commands/cli.py", line 2805, in run
result = ci.function(session, **kw_args)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/save_command/cmd.py", line 79, in provider_save
mgr).save(session, path, **provider_kw)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core_formats/__init__.py", line 79, in save
return cxs_save(session, path, **kw)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 826, in save
session.save(output, version=version, include_maps=include_maps)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 583, in save
mgr.discovery(self._state_containers)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 261, in discovery
raise ValueError("error processing: %s: %s" % (_obj_stack(parents, obj), e))
ValueError: error processing: 'isolde residue stepper 1' ->
<chimerax.isolde.navigate.ResidueStepper object at 0x7f42c411fd50> ->
<chimerax.atomic.molobject.Residue object at 0x7f4301f68aa0>: Error while
saving session data for 'isolde residue stepper 1' ->
<chimerax.isolde.navigate.ResidueStepper object at 0x7f42c411fd50> ->
<chimerax.atomic.molobject.Residue object at 0x7f4301f68aa0>
ValueError: error processing: 'isolde residue stepper 1' -> -> : Error while
saving session data for 'isolde residue stepper 1' -> ->
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/session.py", line 261, in discovery
raise ValueError("error processing: %s: %s" % (_obj_stack(parents, obj), e))
See log for complete Python traceback.
> save reopened.cxs
Taking snapshot of stepper: working_2.pdb
> ui tool show "Build Structure"
> build start peptide "custom built" AAEMSPLPPGR -139.0,135.0 -139.0,135.0
> -139.0,135.0 -139.0,135.0 -139.0,135.0 -139.0,135.0 -139.0,135.0
> -139.0,135.0 -139.0,135.0 -139.0,135.0 -139.0,135.0 rotLib Dunbrack
Chain information for custom built #1
---
Chain | Description
A | No description available
> addh #1
Summary of feedback from adding hydrogens to custom built #1
---
notes | No usable SEQRES records for custom built (#1) chain A; guessing
termini instead
Chain-initial residues that are actual N termini: custom built #1/A ALA 1
Chain-initial residues that are not actual N termini:
Chain-final residues that are actual C termini: custom built #1/A ARG 11
Chain-final residues that are not actual C termini:
0 hydrogen bonds
80 hydrogens added
> select #1
158 atoms, 160 bonds, 11 residues, 1 model selected
> show sel
> ui mousemode right "translate selected models"
> ui mousemode right "rotate selected models"
> ui mousemode right "translate selected models"
> save reopened.cxs
Taking snapshot of stepper: working_2.pdb
Restoring stepper: working_2.pdb
opened ChimeraX session
> ui tool show Shell
/opt/UCSF/ChimeraX/lib/python3.7/site-packages/IPython/core/history.py:226:
UserWarning: IPython History requires SQLite, your history will not be saved
warn("IPython History requires SQLite, your history will not be saved")
> isolde start
> set selectionWidth 4
Done loading forcefield
> select clear
> select clear
> select ~protein
16729 atoms, 17062 bonds, 366 pseudobonds, 140 residues, 23 models selected
> select /ak
1238 atoms, 1261 bonds, 72 residues, 1 model selected
> hide #!1 models
> select up
45 atoms, 44 bonds, 2 residues, 1 model selected
> select up
71 atoms, 71 bonds, 4 residues, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 4 residues in model 8.2
> select up
11 atoms, 10 bonds, 1 residue, 1 model selected
> select up
939 atoms, 951 bonds, 60 residues, 1 model selected
> select up
1079 atoms, 1099 bonds, 61 residues, 1 model selected
> select up
1179 atoms, 1198 bonds, 62 residues, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 66 residues in model 8.2
> select up
20 atoms, 19 bonds, 1 residue, 1 model selected
> select up
159 atoms, 161 bonds, 11 residues, 1 model selected
Chain CG, residue 1 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain CG, residue 2 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain CG, residue 3 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain ak, residue 67 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain CG, residue 1 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain CG, residue 2 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain CG, residue 3 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain ak, residue 67 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> delete sel
> delete sel
> delete sel
> delete sel
> select up
99 atoms, 102 bonds, 4 residues, 1 model selected
> select clear
Chain ak, residue 67 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select /ak
1234 atoms, 1257 bonds, 72 residues, 1 model selected
> select up
10 atoms, 9 bonds, 1 residue, 1 model selected
> select up
155 atoms, 157 bonds, 11 residues, 1 model selected
Chain ak, residue 67 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain ak, residue 67 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select /MG
99 atoms, 102 bonds, 4 residues, 1 model selected
> select up
42 atoms, 42 bonds, 1 residue, 1 model selected
> select clear
> select sel&:MG
Nothing selected
> select sel&/MG
21 atoms, 21 bonds, 1 residue, 1 model selected
> select up
42 atoms, 42 bonds, 1 residue, 1 model selected
> select clear
> select up
99 atoms, 102 bonds, 4 residues, 1 model selected
> select clear
> select up
42 atoms, 42 bonds, 1 residue, 1 model selected
> delete sel
> isolde add ligand RAM
Deleted the following atoms from residue RAM CG4: O1, HO1
> ui mousemode right "translate selected atoms"
> clipper set contourSensitivity 0.25
> select clear
> isolde add ligand RAM
Deleted the following atoms from residue RAM MG4: HO1, O1
> delete sel
> isolde add ligand RAM
Deleted the following atoms from residue RAM MG4: HO1, O1
> ui mousemode right "translate selected atoms"
> select clear
> select /ak
1234 atoms, 1257 bonds, 72 residues, 1 model selected
> select up
10 atoms, 9 bonds, 1 residue, 1 model selected
> select up
155 atoms, 157 bonds, 11 residues, 1 model selected
Chain ak, residue 67 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
> select up
15 atoms, 14 bonds, 1 residue, 1 model selected
> select up
155 atoms, 157 bonds, 11 residues, 1 model selected
Chain ak, residue 67 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
Chain ak, residue 67 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
> isolde ~ignore
> save working_3.cxs
Taking snapshot of stepper: working_2.pdb
Restoring stepper: working_2.pdb
opened ChimeraX session
> select /ak
1234 atoms, 1257 bonds, 72 residues, 1 model selected
> select clear
> dssp
> select /AA
981 atoms, 1002 bonds, 48 residues, 1 model selected
> isolde stepto /AB
Multiple residues selected! Going to the first...
> clipper set contourSensitivity 0.25
> select /AB
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AC
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto prev
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> select /AD
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> select /AE
980 atoms, 1001 bonds, 48 residues, 1 model selected
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AF
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> swapaa mousemode sel PRO
Traceback (most recent call last):
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2707, in _start_sim_or_toggle_pause
self.start_sim()
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2724, in start_sim
main_sel = self._last_main_sel = self._get_main_sim_selection()
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2907, in _get_main_sim_selection
raise TypeError('You must select at least one atom from the current '
TypeError: You must select at least one atom from the current working model
prior to starting a simulation!
TypeError: You must select at least one atom from the current working model
prior to starting a simulation!
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2907, in _get_main_sim_selection
raise TypeError('You must select at least one atom from the current '
See log for complete Python traceback.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AG
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AG
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select /AG, ak
2214 atoms, 2258 bonds, 120 residues, 1 model selected
> select /AG
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AH
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AI
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> save working_4.cxs
Taking snapshot of stepper: working_2.pdb
> select /AJ
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AK
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> select /AK
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AL
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AM
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> select /AM
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AN
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde ignore sel
ISOLDE: currently ignoring 2 residues in model 8.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> isolde ~ignore
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AO
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AP
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AQ
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AR
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AS
980 atoms, 1001 bonds, 48 residues, 1 model selected
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AT
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AU
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto prev
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AV
980 atoms, 1001 bonds, 48 residues, 1 model selected
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AW
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /AX
980 atoms, 1001 bonds, 48 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> select /AA
981 atoms, 1002 bonds, 48 residues, 1 model selected
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /BA
766 atoms, 787 bonds, 40 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select /BB
773 atoms, 794 bonds, 41 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
40 atoms, 39 bonds, 3 residues, 1 model selected
> select up
766 atoms, 787 bonds, 40 residues, 1 model selected
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> clipper spotlight radius 13.00
> clipper spotlight radius 14.00
> clipper spotlight radius 15.00
> isolde stepto
> isolde stepto
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> isolde add ligand LMT
place_ligand() was called with use_md_template=True, but no suitable template
was found. This command has been ignored.
Chain ak, residue 67 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain ak, residue 67 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
> isolde ~ignore
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> delete sel
> save working.cxs
Taking snapshot of stepper: working_2.pdb
opened ChimeraX session
> isolde start
> set selectionWidth 4
Done loading forcefield
> isolde stepto
> clipper set contourSensitivity 0.25
> select /AA
1019 atoms, 1041 bonds, 51 residues, 1 model selected
> show #!1 models
> select #1
158 atoms, 160 bonds, 11 residues, 8 models selected
> select #1
158 atoms, 160 bonds, 11 residues, 8 models selected
> close #1
> isolde stepto /BA
Multiple residues selected! Going to the first...
> isolde stepto /BB
Multiple residues selected! Going to the first...
> select clear
> select /BB
773 atoms, 794 bonds, 41 residues, 1 model selected
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
17 atoms, 16 bonds, 1 residue, 1 model selected
> select up
40 atoms, 39 bonds, 3 residues, 1 model selected
> select up
766 atoms, 787 bonds, 40 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> select up
14 atoms, 13 bonds, 1 residue, 1 model selected
> select up
47 atoms, 46 bonds, 4 residues, 1 model selected
> select up
773 atoms, 794 bonds, 41 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
47 atoms, 46 bonds, 4 residues, 1 model selected
> select up
773 atoms, 794 bonds, 41 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
40 atoms, 39 bonds, 3 residues, 1 model selected
> select up
766 atoms, 787 bonds, 40 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
40 atoms, 39 bonds, 3 residues, 1 model selected
> select up
766 atoms, 787 bonds, 40 residues, 1 model selected
> select up
96639 atoms, 98747 bonds, 5194 residues, 1 model selected
Chain ak, residue 67 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
> select clear
> select clear
> select up
17 atoms, 16 bonds, 1 residue, 1 model selected
> select up
40 atoms, 39 bonds, 3 residues, 1 model selected
> select up
766 atoms, 787 bonds, 40 residues, 1 model selected
> select up
96639 atoms, 98747 bonds, 5194 residues, 1 model selected
> select down
766 atoms, 787 bonds, 40 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
40 atoms, 39 bonds, 3 residues, 1 model selected
> select up
766 atoms, 787 bonds, 40 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
47 atoms, 46 bonds, 4 residues, 1 model selected
> select up
773 atoms, 794 bonds, 41 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
40 atoms, 39 bonds, 3 residues, 1 model selected
> select up
766 atoms, 787 bonds, 40 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
40 atoms, 39 bonds, 3 residues, 1 model selected
> select up
766 atoms, 787 bonds, 40 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
40 atoms, 39 bonds, 3 residues, 1 model selected
> select up
766 atoms, 787 bonds, 40 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
40 atoms, 39 bonds, 3 residues, 1 model selected
> select up
766 atoms, 787 bonds, 40 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
40 atoms, 39 bonds, 3 residues, 1 model selected
> select up
766 atoms, 787 bonds, 40 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
100 atoms, 99 bonds, 1 residue, 1 model selected
> show sel
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
17 atoms, 16 bonds, 1 residue, 1 model selected
> select up
40 atoms, 39 bonds, 3 residues, 1 model selected
> select up
766 atoms, 787 bonds, 40 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
40 atoms, 39 bonds, 3 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
16 atoms, 15 bonds, 1 residue, 1 model selected
> select up
503 atoms, 512 bonds, 32 residues, 1 model selected
> select up
766 atoms, 787 bonds, 40 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
40 atoms, 39 bonds, 3 residues, 1 model selected
> select up
766 atoms, 787 bonds, 40 residues, 1 model selected
> ui tool show Shell
/opt/UCSF/ChimeraX/lib/python3.7/site-packages/IPython/core/history.py:226:
UserWarning: IPython History requires SQLite, your history will not be saved
warn("IPython History requires SQLite, your history will not be saved")
> select up
7 atoms, 6 bonds, 1 residue, 1 model selected
> select up
38 atoms, 37 bonds, 3 residues, 1 model selected
> select up
771 atoms, 792 bonds, 41 residues, 1 model selected
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
7 atoms, 6 bonds, 1 residue, 1 model selected
> select up
38 atoms, 37 bonds, 3 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
503 atoms, 512 bonds, 32 residues, 1 model selected
> select up
771 atoms, 792 bonds, 41 residues, 1 model selected
> select down
503 atoms, 512 bonds, 32 residues, 1 model selected
> select up
771 atoms, 792 bonds, 41 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
7 atoms, 6 bonds, 1 residue, 1 model selected
> select up
38 atoms, 37 bonds, 3 residues, 1 model selected
> select up
771 atoms, 792 bonds, 41 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
7 atoms, 6 bonds, 1 residue, 1 model selected
> select up
38 atoms, 37 bonds, 3 residues, 1 model selected
> select up
771 atoms, 792 bonds, 41 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
63 atoms, 60 bonds, 3 residues, 1 model selected
> select up
1073 atoms, 1092 bonds, 67 residues, 1 model selected
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
40 atoms, 39 bonds, 3 residues, 1 model selected
> select up
766 atoms, 787 bonds, 40 residues, 1 model selected
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
47 atoms, 46 bonds, 4 residues, 1 model selected
> select up
773 atoms, 794 bonds, 41 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
17 atoms, 16 bonds, 1 residue, 1 model selected
> select up
47 atoms, 46 bonds, 4 residues, 1 model selected
> select up
773 atoms, 794 bonds, 41 residues, 1 model selected
> select clear
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> isolde stepto
> select up
9 atoms, 8 bonds, 1 residue, 1 model selected
> select up
40 atoms, 39 bonds, 3 residues, 1 model selected
> select up
766 atoms, 787 bonds, 40 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> select clear
> isolde stepto /aa
Multiple residues selected! Going to the first...
> select up
24 atoms, 23 bonds, 1 residue, 1 model selected
> select up
86 atoms, 87 bonds, 4 residues, 1 model selected
> select up
1062 atoms, 1082 bonds, 60 residues, 1 model selected
> select clear
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
17 atoms, 17 bonds, 1 residue, 1 model selected
> select up
84 atoms, 86 bonds, 4 residues, 1 model selected
> select up
1079 atoms, 1099 bonds, 61 residues, 1 model selected
> select up
96659 atoms, 98767 bonds, 5198 residues, 1 model selected
> select down
1079 atoms, 1099 bonds, 61 residues, 1 model selected
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> view /ab:2
> select clear
> isolde stepto
> view /ab:2
> select clear
> select clear
> isolde stepto
> select up
19 atoms, 18 bonds, 1 residue, 1 model selected
> select up
84 atoms, 86 bonds, 4 residues, 1 model selected
> select up
1079 atoms, 1099 bonds, 61 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> delete sel
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> select up
19 atoms, 18 bonds, 1 residue, 1 model selected
> select up
1079 atoms, 1099 bonds, 61 residues, 1 model selected
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> select clear
> select up
19 atoms, 18 bonds, 1 residue, 1 model selected
> select up
1079 atoms, 1099 bonds, 61 residues, 1 model selected
> select up
96615 atoms, 98722 bonds, 5194 residues, 1 model selected
> select down
1079 atoms, 1099 bonds, 61 residues, 1 model selected
> select clear
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> select up
19 atoms, 18 bonds, 1 residue, 1 model selected
> select up
1079 atoms, 1099 bonds, 61 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde add ligand CDL
place_ligand() was called with use_md_template=True, but no suitable template
was found. This command has been ignored.
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> isolde ignore ~sel
ISOLDE: currently ignoring 5194 residues in model 8.2
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> select down
2 atoms, 1 bond, 1 residue, 1 model selected
> isolde ~ignore
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
> isolde ~ignore
Chain ak, residue 67 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> clipper isolate sel contextDistance 0 maskRadius 3
> select clear
> ~cartoon
> hide HC
> hide #!8.1.1.2 models
> show #!8.1.1.2 models
> select clear
> save resolved_cdl.jpg
> cartoon
> select /ae:CDL
256 atoms, 255 bonds, 1 residue, 1 model selected
> select /ae:11-30@CA
20 atoms, 20 residues, 1 model selected
> select /af:11-30@CA
20 atoms, 20 residues, 1 model selected
> save working_cdl.cxs
Taking snapshot of stepper: working_2.pdb
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> isolde ignore ~sel
ISOLDE: currently ignoring 5195 residues in model 8.2
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 5196 residues in model 8.2
Traceback (most recent call last):
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2707, in _start_sim_or_toggle_pause
self.start_sim()
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2727, in start_sim
self.params, self.sim_params, excluded_residues = self.ignored_residues)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/openmm/openmm_interface.py", line 590, in __init__
self._prepare_mdff_managers()
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/openmm/openmm_interface.py", line 869, in
_prepare_mdff_managers
focus = False)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/symmetry.py", line 982, in
isolate_and_cover_selection
extra_padding=extra_padding)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/maps/map_mgr.py", line 364, in cover_atoms
zm.set_symmetry_map(atoms, transforms, transform_indices)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/maps/mask_handler.py", line 119, in set_symmetry_map
self.structure = self._unique_structure(atoms)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/maps/mask_handler.py", line 185, in
_unique_structure
raise TypeError('All atoms for zone mask must be from a single model!')
TypeError: All atoms for zone mask must be from a single model!
TypeError: All atoms for zone mask must be from a single model!
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/maps/mask_handler.py", line 185, in
_unique_structure
raise TypeError('All atoms for zone mask must be from a single model!')
See log for complete Python traceback.
> isolde ~ignore
> color bychain
> color byhetero
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> isolde ~ignore
> isolde ignore ~sel
ISOLDE: currently ignoring 5195 residues in model 8.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> isolde ~ignore
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select /ag:11-30@CA
20 atoms, 20 residues, 1 model selected
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> isolde ignore ~sel
ISOLDE: currently ignoring 5196 residues in model 8.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> isolde ~ignore
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
> select clear
> isolde ~ignore
> select clear
> save working_cdl.cxs
Taking snapshot of stepper: working_2.pdb
> select /ah:11-30@CA
20 atoms, 20 residues, 1 model selected
> select /AH
1018 atoms, 1040 bonds, 51 residues, 1 model selected
> cartoon
> select :CDL
768 atoms, 765 bonds, 3 residues, 1 model selected
> select /ae, af, ag
4005 atoms, 4062 bonds, 186 residues, 1 model selected
> select /ae, af, ag,ah
5084 atoms, 5161 bonds, 247 residues, 1 model selected
> select clear
> select /ah:11-30@CA
20 atoms, 20 residues, 1 model selected
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> delete sel
> select /ad
1079 atoms, 1099 bonds, 61 residues, 1 model selected
> select /ad,ae
2414 atoms, 2453 bonds, 123 residues, 1 model selected
> select /ad:11-30@CA
20 atoms, 20 residues, 1 model selected
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> delete sel
> select clear
> isolde stepto
> select clear
> select up
24 atoms, 23 bonds, 1 residue, 1 model selected
> select up
84 atoms, 86 bonds, 4 residues, 1 model selected
> select up
184 atoms, 185 bonds, 5 residues, 1 model selected
> select up
2037 atoms, 2081 bonds, 102 residues, 1 model selected
> select down
184 atoms, 185 bonds, 5 residues, 1 model selected
> select up
2037 atoms, 2081 bonds, 102 residues, 1 model selected
> select down
184 atoms, 185 bonds, 5 residues, 1 model selected
> select up
2037 atoms, 2081 bonds, 102 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
17 atoms, 17 bonds, 1 residue, 1 model selected
> select up
1018 atoms, 1040 bonds, 51 residues, 1 model selected
> select clear
> select clear
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select up
17 atoms, 16 bonds, 1 residue, 1 model selected
> select up
1004 atoms, 1025 bonds, 50 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> select clear
> isolde stepto
> select up
17 atoms, 16 bonds, 1 residue, 1 model selected
> select up
1004 atoms, 1025 bonds, 50 residues, 1 model selected
> select clear
> usage isolde jupm
Subcommands are:
* isolde add ligand
* isolde add water
* isolde adjust distances
* isolde adjust torsions
* isolde cisflip
* isolde demo
* isolde ignore
* isolde ~ignore
* isolde jumpto
* isolde pepflip
* isolde release distances
* isolde release torsions
* isolde remote rest info
* isolde remote rest start
* isolde remote rest stop
* isolde remote xmlrpc
* isolde replace ligand
* isolde report
* isolde reset forcefield
* isolde restrain distances
* isolde restrain ligands
* isolde restrain single distance
* isolde restrain torsions
* isolde set
* isolde sim
* isolde start
* isolde stepto
* isolde tutorial
> usage isolde jump
isolde jumpto [direction]
— Jump the view to the first residue of the next chain or the last residue of
the previous chain.
direction: one of next or prev
> isolde jumpto prev
> isolde jumpto prev
> isolde jumpto prev
> isolde jumpto prev
> save working.cxs
Taking snapshot of stepper: working_2.pdb
Restoring stepper: working_2.pdb
opened ChimeraX session
> usage volume mask
volume mask volumes surfaces a surfaces specifier [pad a number] [extend an
integer] [fullMap true or false] [slab slab] [invertMask true or false] [axis
an axis vector] [sandwich true or false] [fillOverlap true or false] [modelId
modelId]
— Mask a map to a surface
slab: a number or some numbers
modelId: a model id
> usage volume zone
volume zone volumes nearAtoms an atoms specifier [range a number]
[bondPointSpacing a number] [minimalBounds true or false] [newMap true or
false] [invert true or false] [subregion map region] [step map step] [modelId
modelId]
— Zero map values beyond a distance range from atoms
modelId: a model id
> volume zone #8.1.1.1 nearAtoms #8 range 1.5 newMap true
> close #1
> select #1
Nothing selected
> select #8
97383 atoms, 99487 bonds, 14 pseudobonds, 5197 residues, 21 models selected
> select clear
> volume zone #8.1.1.1 nearAtoms #8 range 1.5 newMap true
> usage volume subtr
volume subtract volumes [onGrid a density maps specifier] [boundingGrid true
or false] [gridSubregion map region] [gridStep map step] [spacing 1 or 3
floats] [valueType numeric value type] [hideMaps true or false] [subregion map
region] [step map step] [modelId modelId] [inPlace true or false]
[scaleFactors scaleFactors] [minRms true or false]
— Subtract maps pointwise
modelId: a model id
scaleFactors: some numbers
> volume subtract #8.1.1.1 #1 inPlace false
> close #1
> volume #2 level 0.03352
> volume #2 level 0.03748
> select :CDL
768 atoms, 765 bonds, 3 residues, 1 model selected
> select /C1
1472 atoms, 1487 bonds, 92 residues, 1 model selected
> select clear
> delete sel
> select up
12 atoms, 11 bonds, 1 residue, 1 model selected
> select up
125 atoms, 128 bonds, 9 residues, 1 model selected
> hide #!2 models
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> show #!2 models
> select clear
> hide #!2 models
> show #!2 models
> hide #!2 models
> ui tool show "Build Structure"
> build start peptide "custom built" AAAAAAAAAAAAAA -139.0,135.0 -139.0,135.0
> -139.0,135.0 -139.0,135.0 -139.0,135.0 -139.0,135.0 -139.0,135.0
> -139.0,135.0 -139.0,135.0 -139.0,135.0 -139.0,135.0 -139.0,135.0
> -139.0,135.0 -139.0,135.0 rotLib Dunbrack
Chain information for custom built #1
---
Chain | Description
A | No description available
> select #1
71 atoms, 70 bonds, 14 residues, 1 model selected
> ui mousemode right "translate selected models"
> ui mousemode right "rotate selected models"
> ui mousemode right "translate selected models"
> delete /C1:1-14
> addh #1
Summary of feedback from adding hydrogens to custom built #1
---
notes | No usable SEQRES records for custom built (#1) chain A; guessing
termini instead
Chain-initial residues that are actual N termini: custom built #1/A ALA 1
Chain-initial residues that are not actual N termini:
Chain-final residues that are actual C termini: custom built #1/A ALA 14
Chain-final residues that are not actual C termini:
0 hydrogen bonds
72 hydrogens added
> hide #1 models
> select /C1:1-14
143 atoms, 142 bonds, 14 residues, 1 model selected
> select /C1:1-14
143 atoms, 142 bonds, 14 residues, 1 model selected
> swapaa mousemode sel PRO
> select clear
> swapaa mousemode sel PRO
> swapaa mousemode sel GLN
> swapaa mousemode sel THR
> swapaa mousemode sel GLY
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/class1a_24A.mrc
Opened class1a_24A.mrc, grid size 400,400,400, pixel 0.999, shown at level
0.0212, step 2, values float32
> ui tool show "Volume Viewer"
> volume #3 step 1
> volume #3 level 0.04119
> volume #3 level 0.03163
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/class2a_26A.mrc
Opened class2a_26A.mrc, grid size 400,400,400, pixel 0.999, shown at level
0.0249, step 2, values float32
> volume #4 step 1
> volume #4 level 0.05455
> fitmap #4 inMap #8.1.1.1
Fit map class2a_26A.mrc in map class1b_26A.mrc using 135602 points
correlation = 0.0582, correlation about mean = 0.003368, overlap = 2.152
steps = 964, shift = 6.3, angle = 5.34 degrees
Position of class2a_26A.mrc (#4) relative to class1b_26A.mrc (#8.1.1.1)
coordinates:
Matrix rotation and translation
0.99577666 -0.02365016 -0.08871027 25.10161578
0.02499610 0.99958822 0.01409207 -14.07791580
0.08834046 -0.01624996 0.99595778 -13.06451755
Axis -0.16303956 -0.95136249 0.26139533
Axis point 178.35759174 0.00000000 285.28764281
Rotation angle (degrees) 5.33916146
Shift along axis 5.88564083
> fitmap #4 inMap #8.1.1.1
Fit map class2a_26A.mrc in map class1b_26A.mrc using 135602 points
correlation = 0.1152, correlation about mean = 0.06419, overlap = 9.627
steps = 672, shift = 6.89, angle = 21.3 degrees
Position of class2a_26A.mrc (#4) relative to class1b_26A.mrc (#8.1.1.1)
coordinates:
Matrix rotation and translation
0.92701975 -0.37501178 0.00074524 89.89128138
0.37500543 0.92701261 0.00430577 -61.35976247
-0.00230557 -0.00371206 0.99999045 1.56396515
Axis -0.01068950 0.00406739 0.99993459
Axis point 202.60869093 200.28337525 0.00000000
Rotation angle (degrees) 22.02636092
Shift along axis 0.35339585
> select /C1:1-14
159 atoms, 160 bonds, 14 residues, 1 model selected
> select /C1
1784 atoms, 1805 bonds, 117 residues, 1 model selected
> volume #4 level 0.04206
> swapaa mousemode sel GLN
> select up
17 atoms, 16 bonds, 1 residue, 1 model selected
> select up
166 atoms, 167 bonds, 14 residues, 1 model selected
> swapaa mousemode sel ASSP
Missing or invalid "restype" argument: Should be one of 'ALA', 'ARG', 'ASN',
'ASP', 'CYS', 'GLN', 'GLU', 'GLY', 'HIS', 'ILE', 'LEU', 'LYS', 'MET', 'PHE',
'PRO', 'SER', 'THR', 'TRP', 'TYR', or 'VAL'
> swapaa mousemode sel ASP
> swapaa mousemode sel ARG
> swapaa mousemode sel GLY
> swapaa mousemode sel VAL
> swapaa mousemode sel PRO
> select up
16 atoms, 15 bonds, 1 residue, 1 model selected
> select up
190 atoms, 193 bonds, 14 residues, 1 model selected
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Unable to flip peptide bond after 50 rounds. Giving up.
> select /C1:1-14
190 atoms, 193 bonds, 14 residues, 1 model selected
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> select /C1
1815 atoms, 1838 bonds, 117 residues, 1 model selected
> select clear
> select /ak
1234 atoms, 1257 bonds, 72 residues, 1 model selected
> isolde stepto /c
Selection contains no residues!
> isolde stepto /ac
Multiple residues selected! Going to the first...
> select up
17 atoms, 17 bonds, 1 residue, 1 model selected
> select up
84 atoms, 86 bonds, 4 residues, 1 model selected
> select up
1035 atoms, 1054 bonds, 57 residues, 1 model selected
> select clear
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> isolde stepto
> close #1
> show #!2 models
> select :CDL
768 atoms, 765 bonds, 3 residues, 1 model selected
> hide #!2 models
> clipper set contourSensitivity o.25
Missing or invalid "sensitivity" argument: Expected a number
> clipper set contourSensitivity 0.25
> show #!2 models
> select :BCL
11760 atoms, 12432 bonds, 84 residues, 1 model selected
> hide #!2 models
> show #!2 models
> select clear
> hide #!2 models
> select /af
1335 atoms, 1354 bonds, 62 residues, 1 model selected
> select /ag
1335 atoms, 1354 bonds, 62 residues, 1 model selected
> select /ae
1335 atoms, 1354 bonds, 62 residues, 1 model selected
> select down
2 atoms, 1 bond, 1 residue, 1 model selected
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> isolde add ligand CDL
place_ligand() was called with use_md_template=True, but no suitable template
was found. This command has been ignored.
> show #!2 models
> hide #!2 models
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> hide ~sel
> isolde ignore ~sel
ISOLDE: currently ignoring 5222 residues in model 8.2
> show #!2 models
> show ~HC
> hide protein&~@CA
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> select up
1282 atoms, 1300 bonds, 63 residues, 1 model selected
> select down
256 atoms, 255 bonds, 1 residue, 1 model selected
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> select clear
> isolde ~ignore
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> show ~HC
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> isolde ~ignore
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> hide
> hide #!8 models
> volume #2 level 0.03016
> volume #2 level 0.03131
> show #!8 models
> show ~HC
> hide #!8 models
> show #!8 models
> select :CDL
1024 atoms, 1020 bonds, 4 residues, 1 model selected
> hide #!2 models
> show #!2 models
> hide #!2 models
> select clear
> select :CDL
1024 atoms, 1020 bonds, 4 residues, 1 model selected
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> save cdl_ref.pdb #8 selectedOnly true
> open cdl_ref.pdb
Summary of feedback from opening cdl_ref.pdb
---
warnings | Start residue of secondary structure not found: HELIX 1 1 HIS A 2
TRP A 5 1 4
Start residue of secondary structure not found: HELIX 2 2 PRO A 10 GLN A 35 1
26
Start residue of secondary structure not found: HELIX 3 3 ALA A 40 LYS A 45 1
6
Start residue of secondary structure not found: HELIX 4 4 ARG A 3 TRP A 5 1 3
Start residue of secondary structure not found: HELIX 5 5 PRO A 10 PHE A 36 1
27
267 messages similar to the above omitted
Cannot find LINK/SSBOND residue CYS (95 )
Cannot find LINK/SSBOND residue CYS (98 )
Cannot find LINK/SSBOND residue THR (108 )
Cannot find LINK/SSBOND residue CYS (146 )
Cannot find LINK/SSBOND residue CYS (149 )
25 messages similar to the above omitted
> select #1
256 atoms, 255 bonds, 1 residue, 1 model selected
> ui mousemode right "translate selected models"
> hide #1 models
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> isolde ~ignore
> isolde ignore ~sel
ISOLDE: currently ignoring 5223 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 1
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> show #!2 models
> close #2
> select #8
98238 atoms, 100348 bonds, 14 pseudobonds, 5224 residues, 22 models selected
> select clear
> volume zone #8.1.1.1 nearAtoms #8 dist 1.5 newMap true
Expected a keyword
> volume zone #8.1.1.1 nearAtoms #8 range 1.5 newMap true
> volume subtract #8.1.1.1 #2 inPlace false
> close #2
> volume #5 level 0.03153
> volume #5 color #73d216
> select clear
> hide #!5 models
> show #!5 models
> volume #5 level 0.03261
> select :CDL
1536 atoms, 1530 bonds, 6 residues, 2 models selected
> volume #3 level 0.03461
> volume #5 level 0.03526
> volume #5 level 0.0345
> hide #!5 models
> select :MAN
40 atoms, 40 bonds, 2 residues, 1 model selected
> show #!5 models
> select ~CDL
Expected an objects specifier or a keyword
> select :CDL
1536 atoms, 1530 bonds, 6 residues, 2 models selected
> hide #!5 models
> select :GPC
4000 atoms, 3960 bonds, 40 residues, 1 model selected
> select clear
> select ~protein
18265 atoms, 18944 bonds, 14 pseudobonds, 147 residues, 35 models selected
> select clear
> select clear
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> show sel
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> select clear
> select clear
> show sel
> show sel
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> show sel
> select clear
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> select clear
> select clear
> select clear
> select clear
> show sel
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> select clear
> select clear
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
Chain L, residue 68 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
> select clear
> select clear
> select clear
> select clear
> select up
138 atoms, 139 bonds, 1 residue, 1 model selected
> show sel
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Chain ak, residue 67 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
> isolde ignore ~sel
ISOLDE: currently ignoring 5223 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 1
> isolde ~ignore
> select clear
> isolde ~ignore
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> view /C:234
> swapaa mousemode sel GLN
Traceback (most recent call last):
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2707, in _start_sim_or_toggle_pause
self.start_sim()
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2724, in start_sim
main_sel = self._last_main_sel = self._get_main_sim_selection()
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2907, in _get_main_sim_selection
raise TypeError('You must select at least one atom from the current '
TypeError: You must select at least one atom from the current working model
prior to starting a simulation!
TypeError: You must select at least one atom from the current working model
prior to starting a simulation!
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2907, in _get_main_sim_selection
raise TypeError('You must select at least one atom from the current '
See log for complete Python traceback.
> swapaa mousemode sel ALA
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select up
100 atoms, 99 bonds, 1 residue, 1 model selected
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select up
98238 atoms, 100348 bonds, 5224 residues, 1 model selected
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> close #5
> select #8
98238 atoms, 100348 bonds, 14 pseudobonds, 5224 residues, 22 models selected
> select clear
> volume zone #8.1.1.1.1 nearAtoms #8 range 1.5 newMap true
> volume zone #8.1.1.1 nearAtoms #8 range 1.5 newMap true
> volume subtract #8.1.1.1 #2 inPlace false
> close #2
> volume #5 level 0.04033
> volume #5 level 0.03573
> isolde add ligand LMT
place_ligand() was called with use_md_template=True, but no suitable template
was found. This command has been ignored.
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5224 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 1
Chain L, residue 68 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain ak, residue 67 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select /AG:10-30@CA
21 atoms, 21 residues, 1 model selected
> select /AH:10-30@CA
21 atoms, 21 residues, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> delete sel
> show #!5 models
> select /AG,AH,AI,AJ
4153 atoms, 4242 bonds, 205 residues, 1 model selected
> select /AI:10-30@CA
21 atoms, 21 residues, 1 model selected
> select clear
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> ui mousemode right "translate selected atoms"
> hide #!5 models
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select /AG,AI:LMT
162 atoms, 164 bonds, 2 residues, 1 model selected
> show #!5 models
> select /AG,AJ
2117 atoms, 2162 bonds, 103 residues, 1 model selected
> select /AJ:10-30@CA
21 atoms, 21 residues, 1 model selected
> select /AG:LMT
81 atoms, 82 bonds, 1 residue, 1 model selected
> select /AI:LMT
81 atoms, 82 bonds, 1 residue, 1 model selected
> select /AJ:10-30@CA
21 atoms, 21 residues, 1 model selected
> hide #!5 models
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select clear
> view /AL:LMT
> select clear
> select /AM:10-30@CA
21 atoms, 21 residues, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select /AM:10-30@CA
21 atoms, 21 residues, 1 model selected
> view /AM:LMT
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> view /AO:LMT
> select /AP:10-30@CA
21 atoms, 21 residues, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> view /AP:LMT
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> view /AR:LMT
> select clear
> select /AS:10-30@CA
21 atoms, 21 residues, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> view /AS:LMT
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> view /AU:LMT
> select /AV:10-30@CA
21 atoms, 21 residues, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> view /AV:LMT
> view /AV:LMT
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select clear
> view /AX:LMT
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select /AA:10-30@CA
21 atoms, 21 residues, 1 model selected
> view /AA:LMT
> select clear
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> view /AC:LMT
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select /AD:10-30@CA
21 atoms, 21 residues, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> view /AD:LMT
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> view /AF:LMT
> save working.cxs
Taking snapshot of stepper: working_2.pdb
opened ChimeraX session
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1a/lipid_group.pdb
Summary of feedback from opening
/run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1a/lipid_group.pdb
---
warnings | Start residue of secondary structure not found: HELIX 1 1 HIS A 2
TRP A 5 1 4
Start residue of secondary structure not found: HELIX 2 2 PRO A 10 GLN A 35 1
26
Start residue of secondary structure not found: HELIX 3 3 ALA A 40 LYS A 45 1
6
Start residue of secondary structure not found: HELIX 4 4 ARG A 3 TRP A 5 1 3
Start residue of secondary structure not found: HELIX 5 5 PRO A 10 PHE A 36 1
27
264 messages similar to the above omitted
Cannot find LINK/SSBOND residue CYS (95 )
Cannot find LINK/SSBOND residue CYS (98 )
Cannot find LINK/SSBOND residue THR (108 )
Cannot find LINK/SSBOND residue CYS (146 )
Cannot find LINK/SSBOND residue CYS (149 )
25 messages similar to the above omitted
> view #2
> select #2/AJ:61@C5'
1 atom, 1 residue, 1 model selected
> select #2:61
162 atoms, 164 bonds, 2 residues, 1 model selected
> delete sel
> close #2
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1a/lipid_group.pdb
Summary of feedback from opening
/run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1a/lipid_group.pdb
---
warnings | Start residue of secondary structure not found: HELIX 1 1 HIS A 2
TRP A 5 1 4
Start residue of secondary structure not found: HELIX 2 2 PRO A 10 GLN A 35 1
26
Start residue of secondary structure not found: HELIX 3 3 ALA A 40 LYS A 45 1
6
Start residue of secondary structure not found: HELIX 4 4 ARG A 3 TRP A 5 1 3
Start residue of secondary structure not found: HELIX 5 5 PRO A 10 PHE A 36 1
27
264 messages similar to the above omitted
Cannot find LINK/SSBOND residue CYS (95 )
Cannot find LINK/SSBOND residue CYS (98 )
Cannot find LINK/SSBOND residue THR (108 )
Cannot find LINK/SSBOND residue CYS (146 )
Cannot find LINK/SSBOND residue CYS (149 )
25 messages similar to the above omitted
> delete #2/AJ:61
> isolde start
> set selectionWidth 4
Done loading forcefield
> ui tool show Shell
/opt/UCSF/ChimeraX/lib/python3.7/site-packages/IPython/core/history.py:226:
UserWarning: IPython History requires SQLite, your history will not be saved
warn("IPython History requires SQLite, your history will not be saved")
> select #2
194 atoms, 194 bonds, 2 residues, 1 model selected
> hide #2 models
> hide #!1 models
> select clear
> select clear
> select /AK
1406 atoms, 1428 bonds, 55 residues, 2 models selected
> select /AK:10-30@CA
21 atoms, 21 residues, 1 model selected
> view /AA:PEX
> select /AA:PEX
113 atoms, 112 bonds, 1 residue, 1 model selected
> select up
194 atoms, 194 bonds, 2 residues, 1 model selected
> delete sel
> view /AC:PEX
> select up
113 atoms, 112 bonds, 1 residue, 1 model selected
> select up
194 atoms, 194 bonds, 2 residues, 1 model selected
> delete sel
> view /AE:PEX
> select /AA, AC, AE,
Expected an objects specifier or a keyword
> select /AA, AC, AE
3397 atoms, 3464 bonds, 156 residues, 1 model selected
> select clear
> view /AG:PEX
> select /AG:PEX
113 atoms, 112 bonds, 1 residue, 1 model selected
> delete /AA,AC,AE,AG,AI,AM,AO,AQ,AS,AU,AW:61,62
> view /AB:PEX
> view /AE:PEX
> view /AH:PEX
> view /AB:PEX
> select /AB:PEX,LMT
194 atoms, 194 bonds, 2 residues, 1 model selected
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select /AE:PEX,LMT
194 atoms, 194 bonds, 2 residues, 1 model selected
> view sel
> select clear
> select /AH:PEX,LMT
194 atoms, 194 bonds, 2 residues, 1 model selected
> view sel
> select clear
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde add ligand BCL
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 2 residues in model 2
ISOLDE: currently ignoring 5249 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 1.2
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> delete sel
> isolde add ligand BPH
> ui mousemode right "translate selected atoms"
> isolde add ligand BCL
> ui mousemode right "translate selected atoms"
> select up
141 atoms, 146 bonds, 1 residue, 1 model selected
> delete sel
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> isolde ~ignore
> select clear
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> save extra_bcl.pdb #1 selectedOnly true
> select /AN:PEX,LMT
194 atoms, 194 bonds, 2 residues, 1 model selected
> view sel
> select clear
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 2 residues in model 2
ISOLDE: currently ignoring 5250 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 1.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
> select up
19 atoms, 18 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select /AQ:PEX,LMT
194 atoms, 194 bonds, 2 residues, 1 model selected
> view sel
> select clear
> select /AT:PEX,LMT
194 atoms, 194 bonds, 2 residues, 1 model selected
> view sel
> select clear
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> select /AW:PEX,LMT
194 atoms, 194 bonds, 2 residues, 1 model selected
> view sel
> select clear
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> close #5
> select #8
100821 atoms, 102950 bonds, 14 pseudobonds, 5252 residues, 22 models selected
> select clear
> volume zone #8.1.1.1 nearAtoms #8 range 1.5 newMap true
> volume diff #8.1.1.1 #5 inpl f
Expected a density maps specifier or a keyword
> volume subtract #8.1.1.1 #5 inPlace false
> close #5
> show #2 models
> select #2
194 atoms, 194 bonds, 2 residues, 1 model selected
> select clear
> select #2
194 atoms, 194 bonds, 2 residues, 1 model selected
> hide #!6 models
> show #!6 models
> ui mousemode right "translate selected models"
> select #2:PEX
113 atoms, 112 bonds, 1 residue, 1 model selected
> select clear
> volume #6 level 0.0417
> hide #2 models
> hide #!6 models
> select up
113 atoms, 112 bonds, 1 residue, 1 model selected
> isolde ignore ~sel
ISOLDE: currently ignoring 2 residues in model 2
ISOLDE: currently ignoring 5252 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 1.2
> select clear
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> select up
194 atoms, 194 bonds, 2 residues, 1 model selected
> select /AX:10-30@CA
21 atoms, 21 residues, 1 model selected
> select /AC:PEX,LMT
194 atoms, 194 bonds, 2 residues, 1 model selected
> view sel
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> ui mousemode right "translate selected atoms"
> select /AF:PEX,LMT
275 atoms, 276 bonds, 3 residues, 1 model selected
> view sel
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select /AI:PEX,LMT
194 atoms, 194 bonds, 2 residues, 1 model selected
> view sel
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> isolde ~ignore
> select /AL:PEX,LMT
275 atoms, 276 bonds, 3 residues, 1 model selected
> view sel
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select /AO:PEX,LMT
194 atoms, 194 bonds, 2 residues, 1 model selected
> view sel
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> delete sel
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select /AR:PEX,LMT
275 atoms, 276 bonds, 3 residues, 1 model selected
> view sel
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> delete sel
> select /AU:PEX,LMT
194 atoms, 194 bonds, 2 residues, 1 model selected
> view sel
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> close #6
> select #8
102454 atoms, 104584 bonds, 14 pseudobonds, 5269 residues, 22 models selected
> volume zone #8.1.1.1 nearAtoms #8 range 1.5 newMap true
> volume subtract #8.1.1.1 #5 inPlace false
> close #5
> select clear
> select :LMT
2349 atoms, 2378 bonds, 29 residues, 2 models selected
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> delete sel
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> save working.cxs
Taking snapshot of stepper: working_2.pdb
opened ChimeraX session
> isolde start
> set selectionWidth 4
Done loading forcefield
> select up
162 atoms, 164 bonds, 2 residues, 1 model selected
> ui tool show Shell
/opt/UCSF/ChimeraX/lib/python3.7/site-packages/IPython/core/history.py:226:
UserWarning: IPython History requires SQLite, your history will not be saved
warn("IPython History requires SQLite, your history will not be saved")
> select /BH:10-30@CA
21 atoms, 21 residues, 1 model selected
> select /BH:10-40@CA
31 atoms, 31 residues, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select /BG:10-40@CA
31 atoms, 31 residues, 1 model selected
Loading residue template for PEX from internal database
Chain BH, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BI, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BA, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BB, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BC, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BD, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BE, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BF, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BG, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BH, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BI, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BJ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BK, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BL, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BM, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BN, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BO, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BP, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BQ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BR, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BS, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BT, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BU, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BV, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BW, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BX, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain L, residue 68 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain ak, residue 67 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BH, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BI, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BI, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BJ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BJ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BK, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BK, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BL, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select ~protein
28426 atoms, 29196 bonds, 14 pseudobonds, 265 residues, 39 models selected
> select clear
Chain BL, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BM, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
Chain BM, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BN, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Chain BN, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BO, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Chain BO, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BP, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
Chain BP, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BQ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Chain BQ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BR, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> hide #!6 models
Chain BR, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BS, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Chain BS, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BT, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BT, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BU, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
Chain BU, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BV, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BV, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BW, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Chain BW, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BX, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BA, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BX, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Chain BA, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BB, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BB, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BC, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BC, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BD, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BD, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BE, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Chain BE, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BF, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Chain BE, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BF, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BF, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BG, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BG, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BH, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BH, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BI, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BI, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BJ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Chain BJ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BK, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Chain BK, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BL, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Chain BL, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BM, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BM, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BN, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> close #6
> select #8
108286 atoms, 110488 bonds, 14 pseudobonds, 5341 residues, 22 models selected
> volume zone #8.1.1.1 nearAtoms #8 range 1.5 newMap true
> volume subtract #8.1.1.1 #5 inPlace false
> close #5
> select clear
> hide #!6 models
> show #!6 models
> isolde add ligand PC1
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5341 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> volume #6 level 0.04625
> hide #!6 models
> isolde ~ignore
> select up
142 atoms, 141 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
> isolde ~ignore
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> select clear
Chain L, residue 68 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select up
142 atoms, 141 bonds, 1 residue, 1 model selected
> isolde replace ligand sel P5S
Deleted the following atoms from residue PC1 H11001: H143, H151, H122, C14,
C15, H152, H131, H142, H141, H153, H132, C13, H133
126 atoms were automatically renamed to match the template: C12->CA, C11->CB,
O13->OG, P->P12, H111->HB, H112->HBA, H121->HA, O11->O16, C1->C3, C3->C1,
O21->O37, C21->C38, C22->C39, C23->C40, C24->C41, C25->C42, C26->C43,
C27->C44, C28->C45, C29->C46, C2A->C48, C2B->C49, C2C->C50, C2D->C51,
C2E->C52, C2F->C53, C2G->C54, C2H->C55, C2I->C56, O31->O19, C31->C17,
C32->C20, C33->C21, C34->C22, C35->C23, C36->C24, C37->C25, C38->C26,
C39->C27, C3A->C28, C3B->C29, C3C->C30, C3D->C31, C3E->C32, C3F->C33,
C3G->C34, C3H->C35, C3I->C36, O22->O47, O32->O18, H11->H3, H12->H3A,
H221->H39, H222->H39A, H231->H40, H232->H40A, H241->H41, H242->H41A,
H251->H42, H252->H42A, H261->H43, H262->H43A, H271->H44, H272->H44A,
H281->H45, H282->H45A, H291->H46, H292->H46A, H2A1->H48, H2A2->H48A,
H2B1->H49, H2B2->H49A, H2C1->H50, H2C2->H50A, H2D1->H51, H2D2->H51A,
H2E1->H52, H2E2->H52A, H2F1->H53, H2F2->H53A, H2G1->H54, H2G2->H54A,
H2H1->H55, H2H2->H55A, H31->H1, H32->H1A, H321->H20, H322->H20A, H331->H21,
H332->H21A, H341->H22, H342->H22A, H351->H23, H352->H23A, H361->H24,
H362->H24A, H371->H25, H372->H25A, H381->H26, H382->H26A, H391->H27,
H392->H27A, H3A1->H28, H3A2->H28A, H3B1->H29, H3B2->H29A, H3C1->H30,
H3C2->H30A, H3D1->H31, H3D2->H31A, H3E1->H32, H3E2->H32A, H3F1->H33,
H3F2->H33A, H3G1->H34, H3G2->H34A, H3H1->H35, H3H2->H35A, O12->O15, O14->O13,
H2I1->H56, H2I2->H56A, H2I3->H56B, H3I1->H36, H3I2->H36A, H3I3->H36B
Rebuilt ligand P5S has chiral centres at atoms CA,C2 (highlighted). Since the
current algorithm used to match topologies is not chirality aware, you should
check these sites carefully to ensure they are sensible. If in doubt, it is
best to delete with "del #8.2/H1:1001" and replace with "isolde add ligand
P5S".
> isolde replace ligand sel P5S
Deleted the following atoms from residue P5S H11001: HXT, HO15
Traceback (most recent call last):
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/cmd_line/tool.py", line 275, in execute
cmd.run(cmd_text)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/core/commands/cli.py", line 2805, in run
result = ci.function(session, **kw_args)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/atomic/building/cmd.py", line 44, in replace_residue
chiral_centers = Atoms([a for a in residue.atoms if
residue.ideal_chirality(a.name) != 'N'])
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/atomic/building/cmd.py", line 44, in <listcomp>
chiral_centers = Atoms([a for a in residue.atoms if
residue.ideal_chirality(a.name) != 'N'])
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/atomic/molobject.py", line 652, in ideal_chirality
return f(self.name.encode('utf-8'), atom_name.encode('utf-8'))
KeyError: 'Atom H6 not in mmCIF Chemical Component Dictionary for P5S'
KeyError: 'Atom H6 not in mmCIF Chemical Component Dictionary for P5S'
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/atomic/molobject.py", line 652, in ideal_chirality
return f(self.name.encode('utf-8'), atom_name.encode('utf-8'))
See log for complete Python traceback.
Chain L, residue 68 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
> isolde add ligand P5S
Deleted the following atoms from residue P5S H11002: HO15, HXT
> ui mousemode right "translate selected atoms"
> delete sel
Chain L, residue 68 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> show #!6 models
running convert_amber_files
> isolde add ligand 2Y5
Fetching CCD 2Y5 from http://ligand-expo.rcsb.org/reports/2/2Y5/2Y5.cif
Deleted the following atoms from residue 2Y5 L607: H81, H18, H78
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5342 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> hide #!6 models
> delete sel
> isolde add ligand LMG
Fetching CCD LMG from http://ligand-expo.rcsb.org/reports/L/LMG/LMG.cif
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5342 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> delete sel
> select up
129 atoms, 129 bonds, 1 residue, 1 model selected
> save LMX.pdb #8 selectedOnly true
running convert_amber_files
> isolde ~ignore
> select clear
Failed to add
/run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/ligands/PEE.xml:
Residue template USER_PEE with the same override level 0 already exists.
> isolde add ligand PEE
Deleted the following atoms from residue PEE L608: H83
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5343 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> isolde ~ignore
> select up
133 atoms, 132 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
> select clear
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> isolde ~ignore
> select clear
> select :PEE@H35
1 atom, 1 residue, 1 model selected
> select up
133 atoms, 132 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
> isolde ~ignore
> select clear
> select clear
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde add ligand LMG
> ui mousemode right "translate selected atoms"
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
> isolde ~ignore
> isolde ignore ~sel
ISOLDE: currently ignoring 5346 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
> select clear
> isolde add ligand PEE
Deleted the following atoms from residue PEE H11002: H83
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5347 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> isolde ~ignore
> select up
133 atoms, 132 bonds, 1 residue, 1 model selected
> select clear
> show #!6 models
> hide #!6 models
> isolde add ligand PEE
Deleted the following atoms from residue PEE M704: H83
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5348 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
Chain ak, residue 67 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select #8
109253 atoms, 111453 bonds, 14 pseudobonds, 5349 residues, 22 models selected
> close #6
> volume zone #8.1.1.1 nearAtoms #8 range 1.5 newMap true
> volume subtract #8.1.1.1 #5 new t
Expected a keyword
> volume subtract #8.1.1.1 #5 inPlace true
Can't modify volume in place: class1b_26A.mrc
> volume subtract #8.1.1.1 #5 inPlace false
> close #5
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> isolde add ligand LMT
> hide #!6 models
> ui mousemode right "translate selected atoms"
> isolde add ligand LMT
> ui mousemode right "rotate selected models"
> ui mousemode right "translate selected atoms"
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> ui mousemode right "translate selected atoms"
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> show #!6 models
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> hide #!6 models
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> show #!6 models
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> close #6
> select #8
109739 atoms, 111945 bonds, 14 pseudobonds, 5355 residues, 22 models selected
> select clear
> volume zone #8.1.1.1 nearAtoms #8 range 1.5 newMap true
> volume subtract #8.1.1.1 #5 inPlace false
> close #5
> select ~BCL
Expected an objects specifier or a keyword
> select :BCL
11900 atoms, 12580 bonds, 85 residues, 1 model selected
> select down
2 atoms, 1 bond, 1 residue, 1 model selected
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> select /AG:10-40@CA
31 atoms, 31 residues, 1 model selected
> select /AH:10-40@CA
31 atoms, 31 residues, 1 model selected
Chain AH, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BG, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BH, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
Chain AH, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> ui mousemode right "translate selected atoms"
Chain AH, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select /AJ:10-40@CA
31 atoms, 31 residues, 1 model selected
Chain BI, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BJ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select /AK:10-40@CA
31 atoms, 31 residues, 1 model selected
Chain AK, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BJ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BK, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select /AM:10-40@CA
31 atoms, 31 residues, 1 model selected
Chain BL, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BM, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BN, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
> select /AN:10-40@CA
31 atoms, 31 residues, 1 model selected
Chain AN, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BM, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BN, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select /AP:10-40@CA
31 atoms, 31 residues, 1 model selected
Chain BO, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BP, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select /AQ:10-40@CA
31 atoms, 31 residues, 1 model selected
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
Chain AQ, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BQ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
> select /AS:10-40@CA
31 atoms, 31 residues, 1 model selected
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
Chain BS, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
> select /AT:10-40@CA
31 atoms, 31 residues, 1 model selected
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
Chain AT, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BS, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BT, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
> select /AV:10-40@CA
31 atoms, 31 residues, 1 model selected
> hide #!6 models
Chain BU, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BV, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
> show #!6 models
Chain BV, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BW, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select /AW:10-40@CA
31 atoms, 31 residues, 1 model selected
> select ~protein
31499 atoms, 32362 bonds, 14 pseudobonds, 291 residues, 39 models selected
> select clear
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
Chain AW, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BV, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BW, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
> select /AA:10-40@CA
31 atoms, 31 residues, 1 model selected
Chain BA, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BB, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BX, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select /AD:10-40@CA
31 atoms, 31 residues, 1 model selected
Chain BC, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BD, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select /AE:10-40@CA
31 atoms, 31 residues, 1 model selected
Chain AE, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BE, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
Chain AE, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde ignore ~se;
Expected a residues specifier or a keyword
Chain AE, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde ignore ~sel
ISOLDE: currently ignoring 5370 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> isolde ~ignore
Chain AE, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain AH, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BG, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
> select clear
Chain AH, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BG, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> select ~protein
32000 atoms, 32888 bonds, 14 pseudobonds, 295 residues, 39 models selected
> select clear
> hide #!6 models
> hide #!8.1 models
> hide protein
> ~cartoon
> show ~HC
> show #!8.1 models
> cartoon
> select clear
> close #6
> select #8
111861 atoms, 114181 bonds, 14 pseudobonds, 5371 residues, 22 models selected
> select clear
> volume zone #8.1.1.1 nearAtoms #8 range 1.5 newMap true
> volume subtract #8.1.1.1 #5 inPlace false
> close #5
> select clear
> select clear
Chain AW, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BV, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5371 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
Traceback (most recent call last):
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2707, in _start_sim_or_toggle_pause
self.start_sim()
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2727, in start_sim
self.params, self.sim_params, excluded_residues = self.ignored_residues)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/openmm/openmm_interface.py", line 590, in __init__
self._prepare_mdff_managers()
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/openmm/openmm_interface.py", line 869, in
_prepare_mdff_managers
focus = False)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/symmetry.py", line 982, in
isolate_and_cover_selection
extra_padding=extra_padding)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/maps/map_mgr.py", line 364, in cover_atoms
zm.set_symmetry_map(atoms, transforms, transform_indices)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/maps/mask_handler.py", line 119, in set_symmetry_map
self.structure = self._unique_structure(atoms)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/maps/mask_handler.py", line 185, in
_unique_structure
raise TypeError('All atoms for zone mask must be from a single model!')
TypeError: All atoms for zone mask must be from a single model!
TypeError: All atoms for zone mask must be from a single model!
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/maps/mask_handler.py", line 185, in
_unique_structure
raise TypeError('All atoms for zone mask must be from a single model!')
See log for complete Python traceback.
> isolde ~ignore
> show ~HC
> color bychain
> color byhetero
> cartoon
Chain AW, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BV, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BW, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5372 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
Chain AW, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BW, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
Chain AW, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BV, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BW, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde ~ignore
Chain AW, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BV, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BW, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select :LMT
9072 atoms, 9184 bonds, 112 residues, 1 model selected
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5373 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
Chain AT, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BT, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde ~ignore
Chain AT, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde ~ignore
> isolde ignore ~sel
ISOLDE: currently ignoring 5374 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
Chain AT, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select ~protein
32324 atoms, 33216 bonds, 14 pseudobonds, 299 residues, 39 models selected
> select clear
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5375 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> isolde ~ignore
Chain AQ, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
Chain AN, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BM, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BN, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5376 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> isolde ~ignore
Chain AN, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BN, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain AN, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BL, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BM, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5377 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> isolde ~ignore
Chain AN, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BM, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5378 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
Chain AK, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde ~ignore
Chain AK, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BK, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> isolde ignore ~sel
ISOLDE: currently ignoring 5378 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
Chain AK, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BI, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BJ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5379 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> isolde ~ignore
Chain AK, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BJ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5380 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> isolde ~ignore
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
Chain AH, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BH, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BI, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde ~ignore
Chain BH, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BI, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5381 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> isolde ~ignore
Chain AH, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain AH, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde add ligand LMT
> ui mousemode right "rotate selected models"
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5382 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
Chain AE, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> isolde ignore ~sel
ISOLDE: currently ignoring 5382 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> isolde ignore ~sel
ISOLDE: currently ignoring 5382 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5383 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
Chain AE, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BC, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BD, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain AE, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BC, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BD, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> setattr #8 bonds radius 0.05
Assigning radius attribute to 115247 items
> setattr #8 bonds radius 0.2
Assigning radius attribute to 115247 items
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> close #6
> select #8
112914 atoms, 115247 bonds, 14 pseudobonds, 5384 residues, 22 models selected
> volume zone #8.1.1.1 nearAtoms #8 range 1.5 newMap true
> volume subtract #8.1.1.1 #5
> close #5
> select clear
Chain AQ, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5384 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
Chain AQ, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> delete sel
Chain AQ, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BQ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde add ligand PEE
Deleted the following atoms from residue PEE AQ63: H83
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5384 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
Chain AQ, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> select ~protein
33267 atoms, 34168 bonds, 14 pseudobonds, 310 residues, 39 models selected
> select clear
> isolde add ligand P5S
Deleted the following atoms from residue P5S M705: HXT, HO15
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5386 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> hide #!6 models
> select up
135 atoms, 134 bonds, 1 residue, 1 model selected
> delete sel
> isolde add ligand PSF
Fetching CCD PSF from http://ligand-expo.rcsb.org/reports/P/PSF/PSF.cif
Deleted the following atoms from residue PSF M705: HO3, HXT
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5386 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> isolde add ligand PSF
Deleted the following atoms from residue PSF ai68: HO3, HXT
> isolde ignore ~sel
ISOLDE: currently ignoring 5387 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> ui mousemode right "translate selected atoms"
> isolde ~ignore
Chain AN, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select up
62 atoms, 61 bonds, 1 residue, 1 model selected
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> show #!6 models
Chain AH, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain AK, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BJ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> isolde ignore ~sel
ISOLDE: currently ignoring 5387 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
> isolde ~ignore
> isolde ignore se
Expected a residues specifier or a keyword
Chain AH, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain AK, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BJ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
Chain AH, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain AK, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BJ, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde ~ignore
Chain AH, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain AK, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5388 residues in model 8.2
ISOLDE: currently ignoring 1 residues in model 2
ISOLDE: currently ignoring 1 residues in model 1.2
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde ~ignore
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> select ~protein
33472 atoms, 34372 bonds, 14 pseudobonds, 313 residues, 39 models selected
> select clear
Chain AN, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain AN, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
Chain AW, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BA, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
Chain BX, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
Chain AH, residue 62 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> save working.cif #8
Not saving entity_poly_seq for non-authoritative sequences
> select #1
256 atoms, 255 bonds, 1 residue, 8 models selected
> select #8
113333 atoms, 115665 bonds, 14 pseudobonds, 5389 residues, 22 models selected
> select clear
> view sel
> select /ak:67&~protein
140 atoms, 148 bonds, 1 residue, 1 model selected
> view sel
> select clear
> view sel
> select clear
> view sel
> select clear
> view sel
> select clear
> view sel
> select clear
> view sel
> select clear
> view sel
> select clear
> view sel
> select clear
> view sel
> select clear
> view sel
> select clear
> view sel
> view sel
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> save working.cif #8
Not saving entity_poly_seq for non-authoritative sequences
> select ~H
55527 atoms, 116032 bonds, 14 pseudobonds, 5391 residues, 39 models selected
> save working_noh.cif #8 selectedOnly true
Not saving entity_poly_seq for non-authoritative sequences
> select clear
> save working.pdb #8
> view /CG:1
> select /C
4865 atoms, 4968 bonds, 4 pseudobonds, 303 residues, 2 models selected
> select up
78 atoms, 81 bonds, 4 residues, 1 model selected
> view /MG:MAN
> select up
78 atoms, 81 bonds, 4 residues, 1 model selected
> save working.cif #8
Not saving entity_poly_seq for non-authoritative sequences
> select clear
> hide #!6 models
> select up
78 atoms, 81 bonds, 4 residues, 1 model selected
> save working.cif #8
Not saving entity_poly_seq for non-authoritative sequences
> ui mousemode right "translate selected atoms"
> select /C:1010-1020
78 atoms, 81 bonds, 4 residues, 1 model selected
> delete sel
> select /C:1010-1020
78 atoms, 81 bonds, 4 residues, 1 model selected
> delete sel
> select /M:20-50
435 atoms, 440 bonds, 31 residues, 1 model selected
> select /D:20-50
Nothing selected
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1a/model1a.cif
Summary of feedback from opening
/run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1a/model1a.cif
---
warnings | Unknown polymer entity '1' near line 475
Unknown polymer entity '2' near line 2593
Unknown polymer entity '3' near line 5781
Unknown polymer entity '4' near line 26370
Unknown polymer entity '5' near line 27136
10 messages similar to the above omitted
Atom H is not in the residue template for MET /AA:1
Atom C1 is not in the residue template for GPC /AA:60
Atom H is not in the residue template for MET /AB:1
Atom C1 is not in the residue template for GPC /AB:60
Atom H is not in the residue template for MET /AC:1
Atom C1 is not in the residue template for GPC /AC:60
Atom H is not in the residue template for MET /AD:1
Atom C1 is not in the residue template for GPC /AD:60
Atom H is not in the residue template for MET /AE:1
Atom C1 is not in the residue template for GPC /AE:60
Atom H is not in the residue template for MET /AF:1
Atom C1 is not in the residue template for GPC /AF:60
Atom H is not in the residue template for MET /AG:1
Atom C1 is not in the residue template for GPC /AG:60
Atom H is not in the residue template for MET /AH:1
Atom C1 is not in the residue template for GPC /AH:60
Atom H is not in the residue template for MET /AI:1
Atom C1 is not in the residue template for GPC /AI:60
Atom H is not in the residue template for MET /AJ:1
Atom C1 is not in the residue template for GPC /AJ:60
Atom H is not in the residue template for MET /AK:1
Atom C1 is not in the residue template for GPC /AK:60
Atom C26 is not in the residue template for PEX /AK:62
Atom H is not in the residue template for MET /AL:1
Atom C1 is not in the residue template for GPC /AL:60
Atom H is not in the residue template for MET /AM:1
Atom C1 is not in the residue template for GPC /AM:60
Atom H is not in the residue template for MET /AN:1
Atom C1 is not in the residue template for GPC /AN:60
Atom H is not in the residue template for MET /AO:1
Atom C1 is not in the residue template for GPC /AO:60
Atom H is not in the residue template for MET /AP:1
Atom C1 is not in the residue template for GPC /AP:60
Atom H is not in the residue template for MET /AQ:1
Atom C1 is not in the residue template for GPC /AQ:60
Atom H is not in the residue template for MET /AR:1
Atom C1 is not in the residue template for GPC /AR:60
Atom H is not in the residue template for MET /AS:1
Atom C1 is not in the residue template for GPC /AS:60
Atom H is not in the residue template for MET /AT:1
Atom C1 is not in the residue template for GPC /AT:60
Atom H is not in the residue template for MET /AU:1
Atom C1 is not in the residue template for GPC /AU:60
Atom H is not in the residue template for MET /AV:1
Atom C1 is not in the residue template for GPC /AV:60
Atom H is not in the residue template for MET /AW:1
Atom C1 is not in the residue template for GPC /AW:60
Atom H is not in the residue template for MET /AX:1
Atom C1 is not in the residue template for GPC /AX:60
Atom H is not in the residue template for GLY /BQ:5
Atom H is not in the residue template for GLY /BR:5
Atom H is not in the residue template for GLY /BS:5
Atom H is not in the residue template for GLY /BT:5
Atom H11 is not in the residue template for BPH /L:606
Atom H11 is not in the residue template for BPH /M:605
Atom C1 is not in the residue template for RCC /M:701
Atom HN2 is not in the residue template for ARG /ak:71
Atom H is not in the residue template for GLY /C1:32
Atom C1 is not in the residue template for GPC /Ba:60
Atom C1 is not in the residue template for GPC /Bb:60
Atom C1 is not in the residue template for GPC /Bc:60
Atom C1 is not in the residue template for GPC /Bd:60
Atom C1 is not in the residue template for GPC /Be:60
11 messages similar to the above omitted
Missing or incomplete entity_poly_seq table. Inferred polymer connectivity.
Chain information for model1a.cif #5
---
Chain | Description
AA AB AE AF AG AH AI AJ AK AL AM AN AO AP AQ AR AS AT AU AV AW AX | ?
AC AD | ?
BA BC BF BG BH BJ BK BL BM BN BO BP BU BX ba bb bc bd be bf bg bh bi bj bk bl
bm bo bp | ?
BB BD BE BI BQ BR BS BT BV BW bn | ?
C | ?
C1 | ?
H1 | ?
H2 | ?
L | ?
M | ?
aa | ?
ab ad ae af ag ah ai aj al am an ao ap | ?
ac | ?
ak | ?
> cartoon #5
> hide #5
> select /M:20-50
736 atoms, 744 bonds, 2 pseudobonds, 52 residues, 3 models selected
> view /C:1010
No objects specified.
> view /C:1001
> dssp
> select :BCL
25620 atoms, 26748 bonds, 336 pseudobonds, 183 residues, 3 models selected
> close #5
> clipper spotlight radius 13.00
> clipper spotlight radius 14.00
> clipper spotlight radius 15.00
> clipper spotlight radius 16.00
> clipper spotlight radius 17.00
> select /ak
1347 atoms, 1369 bonds, 73 residues, 2 models selected
> view /C:117
> select #8&~protein
33103 atoms, 34005 bonds, 14 pseudobonds, 311 residues, 22 models selected
> select LMG
Expected an objects specifier or a keyword
> select :LMG
141 atoms, 141 bonds, 1 residue, 1 model selected
> view :LMG
> select :LMX
129 atoms, 129 bonds, 1 residue, 1 model selected
> view sel
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
opened ChimeraX session
> show #!6 models
> volume #6 level 0.03545
> ui tool show Shell
/opt/UCSF/ChimeraX/lib/python3.7/site-packages/IPython/core/history.py:226:
UserWarning: IPython History requires SQLite, your history will not be saved
warn("IPython History requires SQLite, your history will not be saved")
> isolde start
> set selectionWidth 4
Done loading forcefield
> select #6
4 models selected
> select :HOH
15 atoms, 10 bonds, 5 residues, 1 model selected
> hide #!6 models
> select :HOH
3309 atoms, 2206 bonds, 1103 residues, 2 models selected
> delete sel
> select :HOH
2505 atoms, 1670 bonds, 835 residues, 1 model selected
> show #!6 models
> hide #!6 models
> delete :HOH
> select :HOH
498 atoms, 332 bonds, 166 residues, 1 model selected
> show #!6 models
> hide #!6 models
> select :HOH
498 atoms, 332 bonds, 166 residues, 1 model selected
> show #!6 models
> delete :HOH
> select :HOH
1497 atoms, 998 bonds, 499 residues, 1 model selected
> hide #!6 models
> show #!6 models
> hide #!6 models
> show #!6 models
> select :HOH
1926 atoms, 1284 bonds, 642 residues, 1 model selected
> hide #!6 models
Loading residue template for LMG from internal database
Loading residue template for PEE from internal database
Loading residue template for PEX from internal database
Loading residue template for PSF from internal database
> select :HOH
1926 atoms, 1284 bonds, 642 residues, 1 model selected
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
> select clear
> select clear
> select clear
> select :HOH
1926 atoms, 1284 bonds, 642 residues, 1 model selected
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
> select clear
> select #8.1.1.1
3 models selected
> select clear
> select :HOH
2085 atoms, 1390 bonds, 695 residues, 1 model selected
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
> select clear
> close #6
> select #1
256 atoms, 255 bonds, 1 residue, 8 models selected
Traceback (most recent call last):
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2580, in _xtal_mask_to_selection
self._xtal_mask_to_atoms(sel, focus)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2589, in _xtal_mask_to_atoms
atoms, 0, context, cutoff, focus=focus)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/symmetry.py", line 982, in
isolate_and_cover_selection
extra_padding=extra_padding)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/maps/map_mgr.py", line 364, in cover_atoms
zm.set_symmetry_map(atoms, transforms, transform_indices)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/maps/mask_handler.py", line 119, in set_symmetry_map
self.structure = self._unique_structure(atoms)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/maps/mask_handler.py", line 185, in
_unique_structure
raise TypeError('All atoms for zone mask must be from a single model!')
TypeError: All atoms for zone mask must be from a single model!
TypeError: All atoms for zone mask must be from a single model!
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/maps/mask_handler.py", line 185, in
_unique_structure
raise TypeError('All atoms for zone mask must be from a single model!')
See log for complete Python traceback.
> select #8
115418 atoms, 117055 bonds, 14 pseudobonds, 6084 residues, 22 models selected
> select clear
> volume zone #8.1.1.1 nearAtoms #8 range 1.5 newMap true
> volume subtract #8.1.1.1 #5
> close #5
> select clear
> select clear
> select :HOH
2085 atoms, 1390 bonds, 695 residues, 1 model selected
> select clear
> hide #!6 models
> show #!6 models
> select clear
> select #6
4 models selected
> select :HOH
1494 atoms, 996 bonds, 498 residues, 1 model selected
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
> hide #!6 models
> select clear
> select :HOH
1494 atoms, 996 bonds, 498 residues, 1 model selected
> isolde add water
> isolde sim start sel
> select :HOH
1095 atoms, 730 bonds, 365 residues, 1 model selected
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> delete sel
> delete sel
> isolde add water
> isolde sim start sel
> isolde add water
> isolde sim start sel
> isolde add water
> isolde sim start sel
> isolde add water
> isolde sim start sel
> delete sel
> setattr sel atoms name H
Assigning name attribute to 1 item
> isolde add water
> isolde sim start sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> isolde add water
> isolde sim start sel
> isolde add water
> isolde sim start sel
> isolde add water
> isolde sim start sel
> isolde add water simSettle false
> isolde add water simSettle false
> show #!6 models
> hide #!6 models
> show #!6 models
> isolde add water
> isolde sim start sel
> hide #!6 models
> isolde add water
> isolde sim start sel
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water
> isolde sim start sel
> isolde add water
> isolde sim start sel
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water
> isolde sim start sel
> isolde add water
> isolde sim start sel
> show #!6 models
> hide #!6 models
> show #!6 models
> hide #!6 models
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water
> isolde sim start sel
> show #!6 models
> volume #6 level 0.02957
> volume #6 level 0.02196
> volume #6 level 0.02872
> hide #!6 models
> isolde add water
> isolde sim start sel
> isolde add water
> isolde sim start sel
> isolde add water simSettle false
> isolde add water simSettle false
> select /ak
1380 atoms, 1391 bonds, 84 residues, 2 models selected
> isolde add water simSettle false
> select /ak
1380 atoms, 1391 bonds, 84 residues, 2 models selected
> select up
114625 atoms, 116562 bonds, 5784 residues, 2 models selected
> select clear
> isolde add water simSettle false
> select /ak
1383 atoms, 1393 bonds, 85 residues, 2 models selected
> select /ak
1383 atoms, 1393 bonds, 85 residues, 2 models selected
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> select clear
> isolde add water simSettle false
> select clear
> view /aa:45
> view /ab:45
> isolde add water simSettle false
> isolde add water simSettle false
> view /ac:45
> select clear
> view /ad:45
> view /ae:45
> view /af:45
> view /ag:45
> view /ah:45
> isolde add water simSettle false
> select clear
> view /ai:45
> view /aj:45
> view /ak:45
> view /al:45
> isolde add water simSettle false
> isolde add water simSettle false
> select clear
> view /am:45
> isolde add water simSettle false
> select clear
> view /an:45
> isolde add water simSettle false
> select clear
> view /ao:45
> view /ap:45
> isolde add water simSettle false
> select clear
> view /aa:5
> view /AA:45
> view /AB:4
> view /AB:43
> view /AC:43
> view /AD:43
> view /AE:43
> select clear
> view /AF:43
> view /AG:43
> view /AH:43
> view /AI:43
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> close #8.1
> clipper associate #3 toModel #8
> volume gaussian #8 bfactor 50
> clipper associate #3 toModel #1
> close #1.1
> volume gaussian #8 bfactor 50
> clipper associate #3 toModel #8
> clipper set contourSensitivity 0.25
> select /D1
Nothing selected
> delete /C2
> select /H1
1580 atoms, 1586 bonds, 75 residues, 1 model selected
> select /M
6670 atoms, 6741 bonds, 6 pseudobonds, 404 residues, 2 models selected
> select clear
> isolde restrain distances #8
> select :HOH
1218 atoms, 812 bonds, 406 residues, 1 model selected
> select /M
6670 atoms, 6741 bonds, 6 pseudobonds, 404 residues, 2 models selected
> select clear
> select /M:20-50
435 atoms, 440 bonds, 31 residues, 1 model selected
> isolde release distances sel
> select clear
> select #8
112951 atoms, 114855 bonds, 14 pseudobonds, 5695 residues, 22 models selected
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
> select clear
> close #8.2.10
> select clear
> select /M:20-50
435 atoms, 440 bonds, 31 residues, 1 model selected
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1a/model1a.cif
Summary of feedback from opening
/run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1a/model1a.cif
---
warnings | Unknown polymer entity '1' near line 475
Unknown polymer entity '2' near line 2593
Unknown polymer entity '3' near line 5781
Unknown polymer entity '4' near line 26370
Unknown polymer entity '5' near line 27136
10 messages similar to the above omitted
Atom H is not in the residue template for MET /AA:1
Atom C1 is not in the residue template for GPC /AA:60
Atom H is not in the residue template for MET /AB:1
Atom C1 is not in the residue template for GPC /AB:60
Atom H is not in the residue template for MET /AC:1
Atom C1 is not in the residue template for GPC /AC:60
Atom H is not in the residue template for MET /AD:1
Atom C1 is not in the residue template for GPC /AD:60
Atom H is not in the residue template for MET /AE:1
Atom C1 is not in the residue template for GPC /AE:60
Atom H is not in the residue template for MET /AF:1
Atom C1 is not in the residue template for GPC /AF:60
Atom H is not in the residue template for MET /AG:1
Atom C1 is not in the residue template for GPC /AG:60
Atom H is not in the residue template for MET /AH:1
Atom C1 is not in the residue template for GPC /AH:60
Atom H is not in the residue template for MET /AI:1
Atom C1 is not in the residue template for GPC /AI:60
Atom H is not in the residue template for MET /AJ:1
Atom C1 is not in the residue template for GPC /AJ:60
Atom H is not in the residue template for MET /AK:1
Atom C1 is not in the residue template for GPC /AK:60
Atom C26 is not in the residue template for PEX /AK:62
Atom H is not in the residue template for MET /AL:1
Atom C1 is not in the residue template for GPC /AL:60
Atom H is not in the residue template for MET /AM:1
Atom C1 is not in the residue template for GPC /AM:60
Atom H is not in the residue template for MET /AN:1
Atom C1 is not in the residue template for GPC /AN:60
Atom H is not in the residue template for MET /AO:1
Atom C1 is not in the residue template for GPC /AO:60
Atom H is not in the residue template for MET /AP:1
Atom C1 is not in the residue template for GPC /AP:60
Atom H is not in the residue template for MET /AQ:1
Atom C1 is not in the residue template for GPC /AQ:60
Atom H is not in the residue template for MET /AR:1
Atom C1 is not in the residue template for GPC /AR:60
Atom H is not in the residue template for MET /AS:1
Atom C1 is not in the residue template for GPC /AS:60
Atom H is not in the residue template for MET /AT:1
Atom C1 is not in the residue template for GPC /AT:60
Atom H is not in the residue template for MET /AU:1
Atom C1 is not in the residue template for GPC /AU:60
Atom H is not in the residue template for MET /AV:1
Atom C1 is not in the residue template for GPC /AV:60
Atom H is not in the residue template for MET /AW:1
Atom C1 is not in the residue template for GPC /AW:60
Atom H is not in the residue template for MET /AX:1
Atom C1 is not in the residue template for GPC /AX:60
Atom H is not in the residue template for GLY /BQ:5
Atom H is not in the residue template for GLY /BR:5
Atom H is not in the residue template for GLY /BS:5
Atom H is not in the residue template for GLY /BT:5
Atom H11 is not in the residue template for BPH /L:606
Atom H11 is not in the residue template for BPH /M:605
Atom C1 is not in the residue template for RCC /M:701
Atom HN2 is not in the residue template for ARG /ak:71
Atom H is not in the residue template for GLY /C1:32
Atom C1 is not in the residue template for GPC /Ba:60
Atom C1 is not in the residue template for GPC /Bb:60
Atom C1 is not in the residue template for GPC /Bc:60
Atom C1 is not in the residue template for GPC /Bd:60
Atom C1 is not in the residue template for GPC /Be:60
11 messages similar to the above omitted
Missing or incomplete entity_poly_seq table. Inferred polymer connectivity.
Chain information for model1a.cif #3
---
Chain | Description
AA AB AE AF AG AH AI AJ AK AL AM AN AO AP AQ AR AS AT AU AV AW AX | ?
AC AD | ?
BA BC BF BG BH BJ BK BL BM BN BO BP BU BX ba bb bc bd be bf bg bh bi bj bk bl
bm bo bp | ?
BB BD BE BI BQ BR BS BT BV BW bn | ?
C | ?
C1 | ?
H1 | ?
H2 | ?
L | ?
M | ?
aa | ?
ab ad ae af ag ah ai aj al am an ao ap | ?
ac | ?
ak | ?
> delete #3&~/M
> select #3/M
5881 atoms, 5992 bonds, 16 pseudobonds, 333 residues, 3 models selected
> style sel stick
Changed 5881 atom styles
> hide HC
> color sel green
> color sel byhetero
> select clear
> select #8/M:20-50
435 atoms, 440 bonds, 31 residues, 1 model selected
> select clear
> hide #!3 models
> select #8/M:20-50
435 atoms, 440 bonds, 31 residues, 1 model selected
> delete #8/M:22-35
> select #8/M:20-50
242 atoms, 244 bonds, 1 pseudobond, 17 residues, 2 models selected
> select #8/M:20-50
242 atoms, 244 bonds, 1 pseudobond, 17 residues, 2 models selected
> isolde add water simSettle false
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> close #6
> close #4
> close #3
> close #2
> close #1
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> select #8
112761 atoms, 114661 bonds, 15 pseudobonds, 5682 residues, 23 models selected
> select clear
> volume zone #8.1.1.1 nearAtoms #8 range 1.5 newMap true
> volume subtract #8.1.1.1 #1
> close #1
> select /M:4-12
162 atoms, 163 bonds, 9 residues, 1 model selected
> select clear
> select up
14 atoms, 13 bonds, 1 residue, 1 model selected
> select up
215 atoms, 216 bonds, 12 residues, 1 model selected
> select clear
> select clear
> select /AA/AX:BCL
1311 atoms, 1345 bonds, 57 residues, 1 model selected
> select /AA-AX:BCL
5460 atoms, 5772 bonds, 39 residues, 1 model selected
> select /AA-AX:BCL
5460 atoms, 5772 bonds, 39 residues, 1 model selected
> isolde add water sel f
Expected a keyword
> isolde add water simSettle false
> select /AA-AX:BCL
5460 atoms, 5772 bonds, 39 residues, 1 model selected
> select /AA-AX:BCL
5460 atoms, 5772 bonds, 39 residues, 1 model selected
> select /AA-AX:BCL
5460 atoms, 5772 bonds, 39 residues, 1 model selected
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> select /AD:10-40@CA
31 atoms, 31 residues, 1 model selected
> select /AB:10-40@CA
31 atoms, 31 residues, 1 model selected
> select /AB
1212 atoms, 1234 bonds, 53 residues, 1 model selected
> select /AA-AX:BCL
5600 atoms, 5920 bonds, 40 residues, 1 model selected
> select clear
> select /AB
1352 atoms, 1382 bonds, 54 residues, 1 model selected
> select /AA-AX:BCL
5600 atoms, 5920 bonds, 40 residues, 1 model selected
> select /AX:10-40@CA
31 atoms, 31 residues, 1 model selected
> select /AX
1302 atoms, 1322 bonds, 57 residues, 1 model selected
> delete /AX:73
> select /AX:10-40@CA
31 atoms, 31 residues, 1 model selected
> select /AA-AX:BCL
5740 atoms, 6068 bonds, 41 residues, 1 model selected
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> select /AA-AX:BCL
5740 atoms, 6068 bonds, 41 residues, 1 model selected
> select /AU:10-40@CA
31 atoms, 31 residues, 1 model selected
> select /AU
1218 atoms, 1238 bonds, 55 residues, 1 model selected
> select /AA-AX:BCL
5880 atoms, 6216 bonds, 42 residues, 1 model selected
> select /AA-AX:BCL
5880 atoms, 6216 bonds, 42 residues, 1 model selected
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> select /AA-AX:BCL
5880 atoms, 6216 bonds, 42 residues, 1 model selected
> select /AR:10-40@CA
31 atoms, 31 residues, 1 model selected
> select /AR
1215 atoms, 1236 bonds, 54 residues, 1 model selected
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select /AA-AX:BCL
6020 atoms, 6364 bonds, 43 residues, 1 model selected
> select /AA-AX:BCL
6020 atoms, 6364 bonds, 43 residues, 1 model selected
> select /AA-AX:BCL
6020 atoms, 6364 bonds, 43 residues, 1 model selected
> select /Ao:10-40@CA
Nothing selected
> select /AO:10-40@CA
31 atoms, 31 residues, 1 model selected
> select /AO
1134 atoms, 1154 bonds, 53 residues, 1 model selected
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> select /AO
1274 atoms, 1302 bonds, 54 residues, 1 model selected
> select /AA-AX:BCL
6160 atoms, 6512 bonds, 44 residues, 1 model selected
> select /AL:10-40@CA
31 atoms, 31 residues, 1 model selected
> select /AL
1305 atoms, 1324 bonds, 58 residues, 1 model selected
> select /AL:10-40@CA
31 atoms, 31 residues, 1 model selected
> select /AA-AX:BCL
6300 atoms, 6660 bonds, 45 residues, 1 model selected
> select clear
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> select /AA-AX:BCL
6300 atoms, 6660 bonds, 45 residues, 1 model selected
> select /AA-AX:BCL
6300 atoms, 6660 bonds, 45 residues, 1 model selected
> select clear
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> delete sel
> select /AF:10-40@CA
31 atoms, 31 residues, 1 model selected
> select /AF
1389 atoms, 1408 bonds, 60 residues, 1 model selected
> select clear
> select /AA-AX:BCL
6440 atoms, 6808 bonds, 46 residues, 1 model selected
> select /AA-AX:BCL
6440 atoms, 6808 bonds, 46 residues, 1 model selected
> select /AI:10-40@CA
31 atoms, 31 residues, 1 model selected
> select /AI
1464 atoms, 1486 bonds, 59 residues, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> delete sel
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select /AA-AX:BCL
6580 atoms, 6956 bonds, 47 residues, 1 model selected
> select /AC:10-40@CA
31 atoms, 31 residues, 1 model selected
> select /AC
1207 atoms, 1225 bonds, 55 residues, 1 model selected
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
> isolde ~ignore
> select /AA-AX:BCL
6720 atoms, 7104 bonds, 48 residues, 1 model selected
> select /AA-AX:BCL
6720 atoms, 7104 bonds, 48 residues, 1 model selected
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select /AA-AX:BCL
6720 atoms, 7104 bonds, 48 residues, 1 model selected
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> select /ba-bp:BCL
2240 atoms, 2368 bonds, 16 residues, 1 model selected
> select /ba-bp,aa-ap:BCL
4480 atoms, 4736 bonds, 32 residues, 1 model selected
> select clear
> select clear
> isolde add water simSettle false
> view /aa:26
> view /ab:26
> select clear
> view /ac:26
> view /ad:26
> view /ae:26
> view /af:26
> view /ag:26
> view /ah:26
> view /ai:26
> view /ap:26
> view /aj:26
> select clear
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> isolde add water simSettle false
> select :HOH
1218 atoms, 812 bonds, 406 residues, 1 model selected
> select #8.1.1.1
3 models selected
> select clear
> close #2
> select #8
113595 atoms, 115653 bonds, 15 pseudobonds, 5601 residues, 23 models selected
> volume zone #8.1.1.1 nearAtoms #8 range 1.5 newMap true
> volume subtract #8.1.1.1 #1
> close #1
> select clear
> select #2
4 models selected
> select clear
> select :HOH
2655 atoms, 1770 bonds, 885 residues, 1 model selected
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
> select clear
> select clear
> select :HOH
2655 atoms, 1770 bonds, 885 residues, 1 model selected
> select clear
> select :HOH
2799 atoms, 1866 bonds, 933 residues, 1 model selected
> select clear
> style sel sphere
Changed 672 atom styles
> select :HOH
2799 atoms, 1866 bonds, 933 residues, 1 model selected
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
> select clear
> select :HOH
2799 atoms, 1866 bonds, 933 residues, 1 model selected
> style sel stick
Changed 2799 atom styles
> select :HOH
2334 atoms, 1556 bonds, 778 residues, 1 model selected
> select clear
> hide #!2 models
> select ~:HOH&~H
55004 atoms, 115015 bonds, 15 pseudobonds, 5282 residues, 27 models selected
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> select up
9 atoms, 6 bonds, 3 residues, 1 model selected
> delete sel
> delete sel
> select up
9 atoms, 6 bonds, 3 residues, 1 model selected
> delete sel
> select #8
114906 atoms, 116527 bonds, 15 pseudobonds, 6038 residues, 23 models selected
> select clear
> select #8
114906 atoms, 116527 bonds, 15 pseudobonds, 6038 residues, 23 models selected
> select clear
> select #8
114906 atoms, 116527 bonds, 15 pseudobonds, 6038 residues, 23 models selected
> select clear
> select clear
> select #8
114906 atoms, 116527 bonds, 15 pseudobonds, 6038 residues, 23 models selected
> select clear
> select up
405 atoms, 410 bonds, 5 residues, 1 model selected
> style sel sphere
Changed 405 atom styles
> style sel ball
Changed 405 atom styles
> style sel stick
Changed 405 atom styles
> color sel lightgreen
> color sel byhetero
> color ~sel lightgrey
> color ~sel byhetero
> color sel green
> color sel byhetero
> select clear
> hide :HOH
> select clear
> save sideways_detergents.jpg
> save sideways_detergents.jpg
> color bychain
> color byhetero
> save working.cxs
Taking snapshot of stepper: working_2.pdb
opened ChimeraX session
> select /aa-ap,ba-bp&~protein
6585 atoms, 6636 bonds, 253 residues, 1 model selected
> isolde start
> set selectionWidth 4
Done loading forcefield
> ui tool show Shell
/opt/UCSF/ChimeraX/lib/python3.7/site-packages/IPython/core/history.py:226:
UserWarning: IPython History requires SQLite, your history will not be saved
warn("IPython History requires SQLite, your history will not be saved")
> select /aa-ap,ba-bp&~protein
6585 atoms, 6636 bonds, 253 residues, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select /aa-ap,ba-bp,AA-AX,BA-BX
89079 atoms, 90638 bonds, 4388 residues, 1 model selected
> select clear
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select /AH
1463 atoms, 1484 bonds, 65 residues, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
Loading residue template for PEE from internal database
Loading residue template for PEX from internal database
Loading residue template for LMG from internal database
Loading residue template for PSF from internal database
> select /aa-ap,ba-bp,AA-AX,BA-BX
89076 atoms, 90636 bonds, 4387 residues, 1 model selected
> select /aa-ap,ba-bp,AA-AX,BA-BX&~protein
30204 atoms, 30692 bonds, 676 residues, 1 model selected
> hide protein
> select /aa-ap,ba-bp&~protein
6501 atoms, 6552 bonds, 251 residues, 1 model selected
> select clear
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select /NA
Nothing selected
> select /AN
1364 atoms, 1390 bonds, 58 residues, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select /AT
1367 atoms, 1392 bonds, 59 residues, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select /AE
1370 atoms, 1394 bonds, 60 residues, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select /aa-ap,ba-bp&~protein
6258 atoms, 6306 bonds, 248 residues, 1 model selected
> select clear
> select /AK
1361 atoms, 1388 bonds, 57 residues, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select up
419 atoms, 426 bonds, 27 residues, 1 model selected
> select /AS
1251 atoms, 1278 bonds, 57 residues, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select /AT
1448 atoms, 1474 bonds, 60 residues, 1 model selected
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> select /aa-ap,ba-bp&~protein&~:HOH
5391 atoms, 5644 bonds, 37 residues, 1 model selected
> select clear
> select /AA-AX,BA-BX&~protein&~:HOH
23574 atoms, 24222 bonds, 226 residues, 1 model selected
> select clear
> select /aa-ap,ba-bp
31156 atoms, 31603 bonds, 1836 residues, 1 model selected
> select /aa-ap,ba-bp&protein
25141 atoms, 25543 bonds, 1591 residues, 1 model selected
> select clear
> select /AA-AX,BA-BX&~protein&~:HOH
23574 atoms, 24222 bonds, 226 residues, 1 model selected
> select zone /aa-ap,ba-bp range 4 & sel
Missing or invalid "range" argument: Expected a number
> select zone /aa-ap,ba-bp range 4
Missing or invalid "range" argument: Expected a number
> select zone /aa-ap,ba-bp 4
Selected 10127 atoms, 2 surfaces
> select sel&/AA-AX,BA-BX&~protein&~:HOH
4211 atoms, 3646 bonds, 114 residues, 1 model selected
> select zone /aa-ap,ba-bp&protein 4
Selected 13233 atoms, 2 surfaces
> select sel&/AA-AX,BA-BX&~protein&~:HOH
3584 atoms, 2961 bonds, 107 residues, 1 model selected
> select sel&:PEX,LMT
2797 atoms, 2354 bonds, 57 residues, 1 model selected
> select up
2638 atoms, 2181 bonds, 57 residues, 1 model selected
> select up
2634 atoms, 2173 bonds, 57 residues, 1 model selected
> select down
2218 atoms, 1914 bonds, 57 residues, 1 model selected
> select up
2560 atoms, 2075 bonds, 57 residues, 1 model selected
> select down
1819 atoms, 1586 bonds, 57 residues, 1 model selected
> show sel
> select up
849 atoms, 659 bonds, 44 residues, 1 model selected
> select up
4076 atoms, 4088 bonds, 44 residues, 1 model selected
> save reassigning_ligands.cxs
Taking snapshot of stepper: working_2.pdb
> select /AA-AX,BA-BX&~protein&~:HOH
19498 atoms, 20134 bonds, 182 residues, 1 model selected
> save reassigning_ligands.cxs
Taking snapshot of stepper: working_2.pdb
Restoring stepper: working_2.pdb
opened ChimeraX session
> select :GPC
4000 atoms, 3960 bonds, 40 residues, 1 model selected
> select /Ba-Bp
1603 atoms, 1586 bonds, 17 residues, 1 model selected
> select /Ba-Bp
Nothing selected
> select /Aa-Ap
Nothing selected
> save reassigning_ligands.cxs
Taking snapshot of stepper: working_2.pdb
Restoring stepper: working_2.pdb
opened ChimeraX session
Traceback (most recent call last):
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/map/volume_viewer.py", line 1530, in <lambda>
QTimer.singleShot(200, lambda f=f: f.setMinimumHeight(50))
RuntimeError: wrapped C/C++ object of type QScrollArea has been deleted
RuntimeError: wrapped C/C++ object of type QScrollArea has been deleted
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/map/volume_viewer.py", line 1530, in
QTimer.singleShot(200, lambda f=f: f.setMinimumHeight(50))
See log for complete Python traceback.
> select ~:HOH
112638 atoms, 115015 bonds, 15 pseudobonds, 5282 residues, 24 models selected
> select zone sel 3
Selected 1994 atoms
> select up
2145 atoms, 1430 bonds, 715 residues, 1 model selected
> select :HOH&~sel
120 atoms, 80 bonds, 40 residues, 1 model selected
> delete sel
> view /L:40
> select up
6 atoms, 4 bonds, 2 residues, 1 model selected
> delete sel
> isolde add ligand MQ9
> delete sel
> isolde add ligand MQ8
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5995 residues in model 8.2
> isolde ~ignore
> save working.cxs
Taking snapshot of stepper: working_2.pdb
> select #8
114902 atoms, 116567 bonds, 15 pseudobonds, 5996 residues, 23 models selected
> show #!2 models
> select /aa-ap
18677 atoms, 18860 bonds, 1123 residues, 1 model selected
> select clear
> select /aa-ap
18677 atoms, 18860 bonds, 1123 residues, 1 model selected
> select clear
> select #8
114902 atoms, 116567 bonds, 15 pseudobonds, 5996 residues, 23 models selected
> select /aa-ap
18677 atoms, 18860 bonds, 1123 residues, 1 model selected
> select clear
> select clear
> select /aa-ap
18677 atoms, 18860 bonds, 1123 residues, 1 model selected
> swapaa mousemode sel MET
> swapaa mousemode sel SER
> swapaa mousemode sel PRO
> swapaa mousemode sel LEU
> swapaa mousemode sel GLY
> delete sel
> hide #!2 models
> select up
14 atoms, 14 bonds, 1 residue, 1 model selected
> select up
159 atoms, 161 bonds, 12 residues, 1 model selected
> swapaa mousemode sel ARG
> swapaa mousemode sel ARG
Traceback (most recent call last):
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/atomic/molarray.py", line 112, in __init__
pointers = numpy.array([i._c_pointer.value for i in items], cptr)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/atomic/molarray.py", line 112, in <listcomp>
pointers = numpy.array([i._c_pointer.value for i in items], cptr)
AttributeError: 'Rotamer' object has no attribute '_c_pointer'
During handling of the above exception, another exception occurred:
Traceback (most recent call last):
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2089, in _commit_rotamer
rrm.commit_preview(rot)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/molobject.py", line 4169, in commit_preview
rr = self.add_restraint(rotamer)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/molobject.py", line 4077, in add_restraint
rr = self._get_restraints(_rotamers([r]), True)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/molobject.py", line 78, in _rotamers
return Rotamers(p)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/molarray.py", line 269, in __init__
super().__init__(c_pointers, Rotamer, Rotamers)
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/atomic/molarray.py", line 117, in __init__
raise ValueError('Collection items of unrecognized type "%s"' % t)
ValueError: Collection items of unrecognized type "<class 'list'>"
ValueError: Collection items of unrecognized type ""
File "/opt/UCSF/ChimeraX/lib/python3.7/site-
packages/chimerax/atomic/molarray.py", line 117, in __init__
raise ValueError('Collection items of unrecognized type "%s"' % t)
See log for complete Python traceback.
No rotamer preview selected! Ignoring command.
> isolde ignore ~sel
ISOLDE: currently ignoring 5994 residues in model 8.2
Traceback (most recent call last):
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2707, in _start_sim_or_toggle_pause
self.start_sim()
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2727, in start_sim
self.params, self.sim_params, excluded_residues = self.ignored_residues)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/openmm/openmm_interface.py", line 590, in __init__
self._prepare_mdff_managers()
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/openmm/openmm_interface.py", line 869, in
_prepare_mdff_managers
focus = False)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/symmetry.py", line 982, in
isolate_and_cover_selection
extra_padding=extra_padding)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/maps/map_mgr.py", line 364, in cover_atoms
zm.set_symmetry_map(atoms, transforms, transform_indices)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/maps/mask_handler.py", line 119, in set_symmetry_map
self.structure = self._unique_structure(atoms)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/maps/mask_handler.py", line 185, in
_unique_structure
raise TypeError('All atoms for zone mask must be from a single model!')
TypeError: All atoms for zone mask must be from a single model!
TypeError: All atoms for zone mask must be from a single model!
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/clipper/maps/mask_handler.py", line 185, in
_unique_structure
raise TypeError('All atoms for zone mask must be from a single model!')
See log for complete Python traceback.
> isolde ~ignore
> color bychain
> color byhetero
> cartoon
> select up
24 atoms, 23 bonds, 1 residue, 1 model selected
> isolde ignore ~sel
ISOLDE: currently ignoring 5993 residues in model 8.2
> isolde ~ignore
Added a C-terminal OXT to chain C1
> select up
24 atoms, 23 bonds, 1 residue, 1 model selected
> show sel
> swapaa sel mousem GLU
Expected a keyword
> swapaa mousemode sel GLU
> swapaa mousemode sel ALA
> swapaa mousemode sel ALA
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> select up
10 atoms, 9 bonds, 1 residue, 1 model selected
> select up
162 atoms, 164 bonds, 12 residues, 1 model selected
> select /ap:50-100
291 atoms, 295 bonds, 23 residues, 1 model selected
Chain ap, residue 60 specifies more than one residue! The simulation can still
run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
> swapaa mousemode sel ALA
> select up
10 atoms, 9 bonds, 1 residue, 1 model selected
> delete sel
> select /ap:50-100
294 atoms, 298 bonds, 23 residues, 1 model selected
> select clear
Residue ALA ap61 is not a C-terminal residue!
Residue ALA ap61 is not a C-terminal residue!
> isolde add ligand TRP
place_ligand() was called with use_md_template=True, but no suitable template
was found. This command has been ignored.
> select up
27 atoms, 28 bonds, 1 residue, 1 model selected
> delete sel
> select :MQ8
125 atoms, 126 bonds, 1 residue, 1 model selected
> select clear
> select /ap
1268 atoms, 1283 bonds, 81 residues, 1 model selected
Added a C-terminal OXT to chain ap
> isolde add water
> isolde sim start sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> style sel stick
Changed 3 atom styles
> color sel byhetero
> select clear
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> select clear
> clipper spotlight radius 19.00
> clipper spotlight radius 20.00
> select up
17 atoms, 16 bonds, 1 residue, 1 model selected
> select up
159 atoms, 161 bonds, 11 residues, 1 model selected
Chain bk, residue 100 specifies more than one residue! The simulation can
still run, but this will probably cause problems later if not rectified by
renumbering.
Deleted the following atoms from residue GLU an63: HN2
Deleted the following atoms from residue SER an65: HN2
Deleted the following atoms from residue LEU an67: HN2
Deleted the following atoms from residue ARG an71: HN2
Chain bk, residue 100 specifies more than one residue! The simulation can
still run, but this will probably cause problems later if not rectified by
renumbering.
> select up
11 atoms, 10 bonds, 1 residue, 1 model selected
> select up
155 atoms, 157 bonds, 11 residues, 1 model selected
> select clear
> show #!2 models
> hide #!2 models
> isolde add ligand LMT chain ba
> ui mousemode right "translate selected atoms"
> select clear
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> select up
9 atoms, 6 bonds, 3 residues, 1 model selected
> select up
12 atoms, 8 bonds, 4 residues, 1 model selected
> delete sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> select up
6 atoms, 4 bonds, 2 residues, 1 model selected
> select up
9 atoms, 6 bonds, 3 residues, 1 model selected
> select up
15 atoms, 10 bonds, 5 residues, 1 model selected
> select up
18 atoms, 12 bonds, 6 residues, 1 model selected
> select up
27 atoms, 18 bonds, 9 residues, 1 model selected
> delete sel
> select clear
> select ~:HOH
112976 atoms, 115358 bonds, 15 pseudobonds, 5293 residues, 27 models selected
> usage sel
select [objects] [residues true or false] [polymer an atoms specifier]
[minimumLength a number] [maximumLength a number] [sequence a text string]
— select specified objects
Subcommands are:
* select add
* select clear
* select down
* select intersect
* select subtract
* select up
* select zone
> select #8.2&~:HOH
112976 atoms, 115358 bonds, 15 pseudobonds, 5293 residues, 13 models selected
> select zone sel 3
Selected 1954 atoms, 5 surfaces
> select up
2103 atoms, 1402 bonds, 701 residues, 6 models selected
> select :HOH&~sel
3 atoms, 2 bonds, 1 residue, 1 model selected
> select clear
> select #8.2&~:HOH
112976 atoms, 115358 bonds, 15 pseudobonds, 5293 residues, 13 models selected
> select zone sel 3
Selected 1954 atoms, 5 surfaces
> select up
2103 atoms, 1402 bonds, 701 residues, 6 models selected
> select down
1954 atoms, 701 residues, 6 models selected
> select clear
> select #8.2&~:HOH&~H
55156 atoms, 115358 bonds, 15 pseudobonds, 5293 residues, 13 models selected
> select zone sel 3
Selected 59310 atoms, 5 surfaces
> select up
114877 atoms, 116626 bonds, 5926 residues, 6 models selected
> select :HOH&~sel
204 atoms, 136 bonds, 68 residues, 1 model selected
> delete sel
> isolde add ligand MQ8
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5927 residues in model 8.2
> isolde ~ignore
Chain bk, residue 100 specifies more than one residue! The simulation can
still run, but this will probably cause problems later if not rectified by
renumbering.
> select clear
> select up
125 atoms, 126 bonds, 1 residue, 1 model selected
> show sel
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Chain bk, residue 100 specifies more than one residue! The simulation can
still run, but this will probably cause problems later if not rectified by
renumbering.
> select :MQ9
138 atoms, 139 bonds, 1 residue, 1 model selected
> select :MQ8,MQ9
388 atoms, 391 bonds, 3 residues, 1 model selected
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> select clear
> select clear
> select up
15 atoms, 10 bonds, 5 residues, 1 model selected
> delete sel
> select clear
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> select clear
> isolde add water simSettle false
> select up
6 atoms, 4 bonds, 2 residues, 1 model selected
> delete sel
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
No existing atoms found within the given distance cutoff of the target
position. You may repeat with a larger cutoff or explicitly specify the
B-factor and chain ID, but keep in mind that placing waters outside of
H-bonding distance to the model is generally inadvisable.
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> select clear
> save working.cxs
Taking snapshot of stepper: working_2.pdb
opened ChimeraX session
> view /M:GCU
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/ligands/glycans/GUX.xml
Opened GUX.xml
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/ligands/glycans/gcx_o_acetyl.pdb
> select #1
29 atoms, 29 bonds, 1 residue, 1 model selected
> ui mousemode right "translate selected atoms"
> select up
2 atoms, 1 bond, 1 residue, 1 model selected
> select up
29 atoms, 29 bonds, 1 residue, 1 model selected
> delete sel
> isolde start
> set selectionWidth 4
Done loading forcefield
> pwd
Current working directory is:
/run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1a
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/ligands/glycans/gcx_o_acetyl.pdb
> select #1
29 atoms, 29 bonds, 1 residue, 1 model selected
> delete #1/M:GCU
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/ligands/glycans/gcx_o_acetyl.pdb
> select #1
29 atoms, 29 bonds, 1 residue, 1 model selected
> delete #8/M:GCU
> ui tool show Shell
/opt/UCSF/ChimeraX/lib/python3.7/site-packages/IPython/core/history.py:226:
UserWarning: IPython History requires SQLite, your history will not be saved
warn("IPython History requires SQLite, your history will not be saved")
> hide #1 models
> select up
67 atoms, 69 bonds, 3 residues, 1 model selected
Chain bk, residue 100 specifies more than one residue! The simulation can
still run, but this will probably cause problems later if not rectified by
renumbering.
> select up
29 atoms, 29 bonds, 1 residue, 1 model selected
> select clear
> select /C
5190 atoms, 5214 bonds, 4 pseudobonds, 390 residues, 2 models selected
> select clear
> view :GUX
> select up
29 atoms, 29 bonds, 1 residue, 1 model selected
> save acetylated_gcu.jpg
> select clear
> view /M:GCU
> view #8:GCU
> view #8:GUX
> close #1#2
> view :GCU
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/ligands/glycans/gcx_o_acetyl.pdb
> select #1
29 atoms, 29 bonds, 1 residue, 1 model selected
> ui mousemode right "translate selected atoms"
> select up
19 atoms, 19 bonds, 1 residue, 1 model selected
> delete sel
> hide #1 models
> select up
67 atoms, 69 bonds, 3 residues, 1 model selected
Chain bk, residue 100 specifies more than one residue! The simulation can
still run, but this will probably cause problems later if not rectified by
renumbering.
> select up
29 atoms, 29 bonds, 1 residue, 1 model selected
> select clear
> save model1a_2020_11_02.pdb #1
> save model1a_2020_11_02.pdb #8
> view /M:GUX
> select clear
> isolde add water simSettle false
> save model1a_2020_11_02.pdb #8
> isolde add water simSettle false
> isolde add water simSettle false
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> isolde add water simSettle false
> isolde add water simSettle false
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> select clear
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> select clear
> isolde add water simSettle false
> isolde add water simSettle false
> select clear
> isolde add water simSettle false
> select clear
> isolde add water simSettle false
> select clear
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> select /a*:50-60
2020 atoms, 2035 bonds, 172 residues, 1 model selected
> select clear
> isolde add water simSettle false
> select clear
> select clear
> select clear
> isolde add water simSettle false
> select clear
> isolde add water simSettle false
> isolde add water simSettle false
> select clear
> isolde add water simSettle false
> select clear
> isolde add water simSettle false
> isolde add water simSettle false
> isolde add water simSettle false
> select up
6 atoms, 4 bonds, 2 residues, 1 model selected
> delete sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> select clear
> save model1a_2020_11_02.cxs
Taking snapshot of stepper: Model 1a
> save model1a_2020_11_02.pdb #8
> select clear
> delete sel
> dssp
> save model1a_2020_11_02.cxs
Taking snapshot of stepper: Model 1a
opened ChimeraX session
> close #8.1.1.2
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2020_10_new_maps/model1a/class1a_r_2.35A.mrc
Opened class1a_r_2.35A.mrc, grid size 400,400,400, pixel 0.999, shown at level
0.0164, step 2, values float32
> clipper associate #2 toModel #8
> close #1
> isolde start
> set selectionWidth 4
Done loading forcefield
> view /M:MAN
> select clear
> clipper spotlight radius 11.00
> clipper spotlight radius 10.00
> clipper spotlight radius 9.00
> clipper spotlight radius 8.00
> clipper spotlight radius 7.00
> clipper spotlight radius 6.00
> clipper spotlight radius 5.00
> clipper spotlight radius 6.00
> clipper spotlight radius 7.00
> clipper spotlight radius 8.00
> clipper spotlight radius 9.00
> clipper spotlight radius 10.00
> clipper spotlight radius 11.00
> clipper spotlight radius 12.00
> clipper spotlight radius 13.00
> clipper spotlight radius 14.00
> clipper spotlight radius 15.00
> clipper spotlight radius 16.00
> select clear
> select clear
> save man.jpg
> select clear
> save model1a_2020_11_04.cxs
Taking snapshot of stepper: Model 1a
opened ChimeraX session
> ui tool show Shell
/opt/UCSF/ChimeraX/lib/python3.7/site-packages/IPython/core/history.py:226:
UserWarning: IPython History requires SQLite, your history will not be saved
warn("IPython History requires SQLite, your history will not be saved")
> open class4b_r_2.35A_postprocess_masked.mrc
Opened class4b_r_2.35A_postprocess_masked.mrc, grid size 400,400,400, pixel
0.999, shown at level 0.0164, step 2, values float32
> volume gaussian #1 bfactor 50
> close #8.1
> clipper associate #1,2 toModel #8
> isolde start
> set selectionWidth 4
Done loading forcefield
> clipper set contourSensitivity 0.25
> select clear
> ui tool show Shell
> alias keep setattr sel residues isolde_keep true
> alias st isolde step $*
> alias aw isolde add water $*
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> select clear
> select clear
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> aw
> isolde sim start sel
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> save working.cxs
Taking snapshot of stepper: Model 1a
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> view /aa:4
> view /aa:47
> view /ab:47
> view /ac:47
> view /ad:47
> view /ae:47
> view /af:47
> view /ag:47
> view /ah:47
> view /ai:47
> view /aj:47
> view /ak:47
> view /al:47
> view /am:47
> view /an:47
> view /ao:47
> view /ap:47
> view /aq:47
No objects specified.
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> setattr sel residues isolde_keep false
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> setattr sel residues isolde_keep false
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> setattr sel residues isolde_keep false
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select up
5218 atoms, 5236 bonds, 396 residues, 1 model selected
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> aw sim f
> aw sim f
> keep
Assigning isolde_keep attribute to 2 items
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> setattr sel residues isolde_keep false
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> setattr sel residues isolde_keep false
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> aw
> isolde sim start sel
> select up
6 atoms, 4 bonds, 2 residues, 1 model selected
> aw
> isolde sim start sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> select up
6 atoms, 4 bonds, 2 residues, 1 model selected
> keep
Assigning isolde_keep attribute to 2 items
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> select up
1257 atoms, 1282 bonds, 59 residues, 1 model selected
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> keep
Assigning isolde_keep attribute to 1 item
> select clear
> select clear
> save working.cxs
Taking snapshot of stepper: Model 1a
> select :HOH
828 atoms, 552 bonds, 276 residues, 1 model selected
> select #1
Nothing selected
> select #8
113879 atoms, 115985 bonds, 15 pseudobonds, 5566 residues, 23 models selected
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
Map is too large for fast cubic interpolation on the GPU! Switching to slower,
more memory-efficient implementation.
> select clear
> select up
215 atoms, 216 bonds, 12 residues, 1 model selected
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> view /ba:32
> view /bb:32
> view /bc:32
> view /bd:32
> view /ba:32
> view /bb:32
> view /bc:32
> view /bd:32
> view /ba:27-32
> view /bb:27-32
> view /bc:27-32
> view /be:27-32
> select clear
> view /bf:27-32
> view /bg:27-32
> view /bh:27-32
> view /bi:27-32
> select clear
> view /bj:27-32
> select clear
> view /bk:27-32
> select clear
> view /bl:27-32
> view /bl:27-32
> view /bm:27-32
> select clear
> select clear
> select clear
> select clear
> view /bn:27-32
> select up
16 atoms, 15 bonds, 1 residue, 1 model selected
> select up
503 atoms, 512 bonds, 32 residues, 1 model selected
> select clear
> select clear
> view /bo:27-32
> select up
16 atoms, 15 bonds, 1 residue, 1 model selected
> select up
503 atoms, 512 bonds, 32 residues, 1 model selected
> select clear
> view /bp:27-32
> select up
16 atoms, 15 bonds, 1 residue, 1 model selected
> select up
503 atoms, 512 bonds, 32 residues, 1 model selected
> select clear
> select clear
> view /bq:27-32
No objects specified.
> save working.cxs
Taking snapshot of stepper: Model 1a
Restoring stepper: Model 1a
opened ChimeraX session
> select :MAN
40 atoms, 40 bonds, 2 residues, 1 model selected
> select clear
> select :XYS
36 atoms, 36 bonds, 2 residues, 1 model selected
> select :XYS
36 atoms, 36 bonds, 2 residues, 1 model selected
> select clear
> movie record
> movie stop
> movie reset
> movie record
> movie stop
> movie encode glycan.mp4
Movie saved to glycan.mp4
> ui tool show "Change Substituents"
> substitute sel substituents COCH3 guessAttachment true modify true minimize
> false
> substitute sel substituents OH guessAttachment true modify true minimize
> false
> substitute sel substituents COCH3 guessAttachment true modify true minimize
> false
> select up
23 atoms, 23 bonds, 1 residue, 1 model selected
> setattr sel residues name XYX
Assigning name attribute to 1 item
> show sel
> setattr sel atoms name O2
Assigning name attribute to 1 item
> select up
23 atoms, 23 bonds, 1 residue, 1 model selected
> show sel
> select up
23 atoms, 23 bonds, 1 residue, 1 model selected
> show sel
> open 3e80
3e80 title:
Structure of Heparinase II complexed with heparan sulfate degradation
disaccharide product [more info...]
Chain information for 3e80 #1
---
Chain | Description
A B C | Heparinase II protein
Non-standard residues in 3e80 #1
---
GCD — 4-deoxy-alpha-L-threo-hex-4-enopyranuronic acid
GCU — alpha-D-glucopyranuronic acid
MAN — alpha-D-mannopyranose
NDG — 2-acetamido-2-deoxy-alpha-D-glucopyranose
PO4 — phosphate ion
RAM — alpha-L-rhamnopyranose
XYS — alpha-D-xylopyranose
ZN — zinc ion
3e80 mmCIF Assemblies
---
1| author_and_software_defined_assembly
2| author_and_software_defined_assembly
3| author_and_software_defined_assembly
> select #1/A:RAM
Nothing selected
> select #1/B:RAM
Nothing selected
> select #1/C:RAM
Nothing selected
> select #1/C:MAN
Nothing selected
> select #1:MAN
33 atoms, 33 bonds, 3 residues, 1 model selected
> select #1/F:MAN
11 atoms, 11 bonds, 1 residue, 1 model selected
> align sel #8/M:MAN&~H
Expected a keyword
> align sel toAtoms #8/M:MAN&~H
RMSD between 11 atom pairs is 0.195 angstroms
> close #1
> open 2fuq
2fuq title:
Crystal Structure of Heparinase II [more info...]
Chain information for 2fuq #1
---
Chain | Description
A B | heparinase II protein
Non-standard residues in 2fuq #1
---
FMT — formic acid
GCU — alpha-D-glucopyranuronic acid
MAN — alpha-D-mannopyranose
PO4 — phosphate ion
RAM — alpha-L-rhamnopyranose
XYS — alpha-D-xylopyranose
ZN — zinc ion
> select #1/C:MAN
11 atoms, 11 bonds, 1 residue, 1 model selected
> align sel toAtoms #8/M:MAN&~H
RMSD between 11 atom pairs is 0.151 angstroms
> hide #!8 models
> show #1
> hide #1&protein
> delete #1&:HOH
> show #!8 models
> hide #!1 models
> select up
23 atoms, 23 bonds, 1 residue, 1 model selected
> select clear
> ui mousemode right "translate selected atoms"
> setattr sel atoms name C2A
Assigning name attribute to 1 item
> select clear
> setattr sel atoms name O2A
Assigning name attribute to 1 item
> setattr sel atoms name C2B
Assigning name attribute to 1 item
> setattr sel atoms name H2B1
Assigning name attribute to 1 item
> setattr sel atoms name H2B2
Assigning name attribute to 1 item
> setattr sel atoms name H2B3
Assigning name attribute to 1 item
> select :XYS
34 atoms, 34 bonds, 3 residues, 2 models selected
> select #8:XYS
18 atoms, 18 bonds, 1 residue, 1 model selected
> view sel
> ui tool show "Change Substituents"
> substitute sel substituents COCH3 newName XYX guessAttachment true modify
> true minimize false
> select clear
> setattr sel atoms name C2A
Assigning name attribute to 1 item
> select up
23 atoms, 23 bonds, 1 residue, 1 model selected
> show sel
> setattr sel atoms name O2A
Assigning name attribute to 1 item
> setattr sel atoms name C2B
Assigning name attribute to 1 item
> setattr sel atoms name H2B1
Assigning name attribute to 1 item
> setattr sel atoms name H2B2
Assigning name attribute to 1 item
> setattr sel atoms name H2B3
Assigning name attribute to 1 item
> select clear
> save working.cxs
Taking snapshot of stepper: Model 1a
> select clear
> select ~H
67529 atoms, 128381 bonds, 21 pseudobonds, 7075 residues, 25 models selected
> save model1a_working_noh.cif #8 selectedOnly true
Not saving entity_poly_seq for non-authoritative sequences
> select clear
> isolde add ligand NDG
Deleted the following atoms from residue NDG M1175: HO1, O1
> ui mousemode right "translate selected atoms"
> show sel
> isolde add ligand A2G
Deleted the following atoms from residue A2G M1176: HO1, O1
> ui mousemode right "translate selected atoms"
> show sel
> delete sel
> select up
23 atoms, 23 bonds, 1 residue, 1 model selected
> delete sel
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> select clear
> view :XYX
> select up
23 atoms, 23 bonds, 1 residue, 1 model selected
> delete sel
> isolde add ligand NDG
Deleted the following atoms from residue NDG C1155: HO1, O1
> show sel
> ui mousemode right "translate selected atoms"
> view /M:NDG
> select ~H
67533 atoms, 128391 bonds, 21 pseudobonds, 7075 residues, 25 models selected
> save model1a_working_noh.cif #8 selectedOnly true
Not saving entity_poly_seq for non-authoritative sequences
> select clear
> save working.cxs
Taking snapshot of stepper: Model 1a
opened ChimeraX session
> close #1
> isolde start
> set selectionWidth 4
Done loading forcefield
> select ~protein&~:HOH
34491 atoms, 35465 bonds, 14 pseudobonds, 320 residues, 21 models selected
> select clear
> st first
> st
> select clear
> st
> st interp 5
> st interp 5
> st
> st
> st
> st
> delete sel
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> st
> st
> st
> st
> st
> awsf
> awsf
> awsf
> awsf
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> st
> st
> st
> st
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> awsf
> awsf
> awsf
> awsf
> awsf
> awsf
> awsf
> awsf
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> awsf
> select clear
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> awsf
> awsf
> st
> st
> st
> aw
> isolde sim start sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> st
> awsf
> awsf
> awsf
> select up
6 atoms, 4 bonds, 2 residues, 1 model selected
> delete sel
> st
> awsf
> awsf
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> awsf
> awsf
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> aw
> isolde sim start sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> aw
> isolde sim start sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> select clear
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> awsf
> st
> st
> aw
> isolde sim start sel
> awsf
> awsf
> select clear
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> awsf
> awsf
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> awsf
> awsf
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> awsf
> select clear
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select up
215 atoms, 216 bonds, 12 residues, 1 model selected
> select clear
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> staw
Unknown command: staw
> aw
> isolde sim start sel
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> awsf
> awsf
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> select clear
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> select clear
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> awsf
> awsf
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> save working.cxs
Taking snapshot of stepper: Model 1a
opened ChimeraX session
> isolde start
> set selectionWidth 4
Done loading forcefield
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> st
> st
> st
> st
> awsf
> awsf
> select clear
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> awsf
> awsf
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> aw
No existing atoms found within the given distance cutoff of the target
position. You may repeat with a larger cutoff or explicitly specify the
B-factor and chain ID, but keep in mind that placing waters outside of
H-bonding distance to the model is generally inadvisable.
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> awsf
> awsf
> select clear
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> select clear
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> select clear
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> aw
> isolde sim start sel
> st
> aw
> isolde sim start sel
> st
> st
> select clear
> st
> st
> st
> awsf
> awsf
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> aw
> isolde sim start sel
> st
> st
> st
> select clear
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> aw
> isolde sim start sel
> st
> st
> aw
> isolde sim start sel
> st
> st
> select clear
> select clear
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> st
> st
> select clear
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> aw
> isolde sim start sel
> st
> select clear
> st
> st
> select clear
> select clear
> aw
> isolde sim start sel
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> st
> st
> st
> st
> awsf
> awsf
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
No existing atoms found within the given distance cutoff of the target
position. You may repeat with a larger cutoff or explicitly specify the
B-factor and chain ID, but keep in mind that placing waters outside of
H-bonding distance to the model is generally inadvisable.
> awst
Unknown command: awst
> st
> st
> st
> awsf
No existing atoms found within the given distance cutoff of the target
position. You may repeat with a larger cutoff or explicitly specify the
B-factor and chain ID, but keep in mind that placing waters outside of
H-bonding distance to the model is generally inadvisable.
> awsf
No existing atoms found within the given distance cutoff of the target
position. You may repeat with a larger cutoff or explicitly specify the
B-factor and chain ID, but keep in mind that placing waters outside of
H-bonding distance to the model is generally inadvisable.
> awsf
> awsf
> st
> st
> select clear
> st
> awsf
> awsf
> st
> st
> select clear
> st
> st
> select clear
> st
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> aw
> isolde sim start sel
> st
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> select clear
> st
> st
> aw
> isolde sim start sel
> select clear
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> select clear
> st
> st
> st
> select up
14 atoms, 14 bonds, 1 residue, 1 model selected
> select up
364 atoms, 368 bonds, 29 residues, 1 model selected
> select clear
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> save working.cxs
Taking snapshot of stepper: Model 1a
> view /ap:30
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> select clear
> st
> aw
> isolde sim start sel
> select clear
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> st
> view /ap:30
> isolde add ligand MQ8
> isolde ignore ~sel
ISOLDE: currently ignoring 5721 residues in model 8.2
> color sel purple
> color sel byhetero
> select /a*:30
320 atoms, 320 bonds, 16 residues, 1 model selected
> select up
125 atoms, 126 bonds, 1 residue, 1 model selected
> isolde ~ignore sel
ISOLDE: currently ignoring 5721 residues in model 8.2
> select up
125 atoms, 126 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 5721 residues in model 8.2
> select up
125 atoms, 126 bonds, 1 residue, 1 model selected
> select clear
> isolde ~ignore
> select up
125 atoms, 126 bonds, 1 residue, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 1 residues in model 8.2
> select up
125 atoms, 126 bonds, 1 residue, 1 model selected
> select clear
> select clear
> select up
270 atoms, 272 bonds, 3 residues, 1 model selected
> clipper isolate sel contextDistance 0
> clipper isolate sel contextDistance 0 maskRadius 7
> clipper isolate sel contextDistance 0 maskRadius 5
> clipper isolate sel contextDistance 0 maskRadius 6
> select sel&~H
117 atoms, 272 bonds, 3 residues, 2 models selected
> save alt_mq8.pdb #8 selectedOnly true
> select up
125 atoms, 126 bonds, 1 residue, 1 model selected
> delete sel
> isolde ~ignore
> select :CDL
1280 atoms, 1275 bonds, 5 residues, 1 model selected
> select clear
> select /a*,b*
37197 atoms, 37702 bonds, 1867 residues, 1 model selected
> select /a*,b*&protein
25462 atoms, 25870 bonds, 1614 residues, 1 model selected
> select :CDL
1280 atoms, 1275 bonds, 5 residues, 1 model selected
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2021_01_final_maps/model1a/alt_mq8.pdb
Summary of feedback from opening
/run/media/tic20/storage/structure_dump/collaboration/pu_qian/2021_01_final_maps/model1a/alt_mq8.pdb
---
warnings | Start residue of secondary structure not found: HELIX 1 1 HIS A 2
TRP A 5 1 4
Start residue of secondary structure not found: HELIX 2 2 PRO A 10 PHE A 36 1
27
Start residue of secondary structure not found: HELIX 3 3 ALA A 40 TYR A 46 1
7
Start residue of secondary structure not found: HELIX 4 4 HIS A 2 TRP A 5 1 4
Start residue of secondary structure not found: HELIX 5 5 PRO A 10 GLN A 35 1
26
248 messages similar to the above omitted
Cannot find LINK/SSBOND residue CYS (95 )
Cannot find LINK/SSBOND residue CYS (98 )
Cannot find LINK/SSBOND residue CYS (146 )
Cannot find LINK/SSBOND residue CYS (149 )
Cannot find LINK/SSBOND residue CYS (216 )
25 messages similar to the above omitted
> view #1
> select #1
117 atoms, 119 bonds, 3 residues, 1 model selected
> close #1
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> aw
> isolde sim start sel
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> delete sel
> st
> st
> st
> st
> select clear
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> st
> save working.cxs
Taking snapshot of stepper: Model 1a
> ui tool show Shell
/opt/UCSF/ChimeraX/lib/python3.7/site-packages/IPython/core/history.py:226:
UserWarning: IPython History requires SQLite, your history will not be saved
warn("IPython History requires SQLite, your history will not be saved")
> save working.cxs
Taking snapshot of stepper: Model 1a
Restoring stepper: Model 1a
opened ChimeraX session
> select clear
> aw
> isolde sim start sel
> isolde jumpto
> select clear
> isolde jumpto
> select clear
> select clear
> aw
> isolde sim start sel
> isolde jumpto
> select clear
> select /AA-AD:1
80 atoms, 76 bonds, 4 residues, 1 model selected
> select clear
Traceback (most recent call last):
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2708, in _start_sim_or_toggle_pause
self.start_sim()
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2728, in start_sim
self.params, self.sim_params, excluded_residues = self.ignored_residues)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/openmm/openmm_interface.py", line 580, in __init__
self._prepare_validation_managers(mobile_atoms)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/openmm/openmm_interface.py", line 753, in
_prepare_validation_managers
rota_a.restrict_to_selected_residues(mobile_res)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/validation/rota_annotation.py", line 129, in
restrict_to_selected_residues
self.update_graphics()
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/validation/rota_annotation.py", line 191, in
update_graphics
non_favored_only = self._hide_favored)
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/molobject.py", line 1660, in
validate_scale_and_color_rotamers
non_favored_only, visible_only, pointer(rot_out), pointer(scale_out),
pointer(color_out))
IndexError: _Map_base::at
IndexError: _Map_base::at
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/molobject.py", line 1660, in
validate_scale_and_color_rotamers
non_favored_only, visible_only, pointer(rot_out), pointer(scale_out),
pointer(color_out))
See log for complete Python traceback.
> save working_fme.cxs
Taking snapshot of stepper: Model 1a
opened ChimeraX session
> isolde start
> set selectionWidth 4
Done loading forcefield
> select clear
> select clear
> st
> isolde jumpto
> ui tool show Shell
/opt/UCSF/ChimeraX/lib/python3.7/site-packages/IPython/core/history.py:226:
UserWarning: IPython History requires SQLite, your history will not be saved
warn("IPython History requires SQLite, your history will not be saved")
> isolde jumpto
> aw
> isolde sim start sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> st
> isolde jumpto
> select clear
> isolde jumpto
> isolde jumpto
> select clear
> isolde jumpto
> isolde jumpto
> isolde jumpto
> aw
> isolde sim start sel
> isolde jumpto
> select clear
> isolde jumpto
> select clear
> select clear
> isolde jumpto
> select clear
> aw
> isolde sim start sel
> isolde jumpto
> select clear
> select clear
> isolde jumpto
> select clear
> isolde jumpto
> awsf
> select clear
> isolde jumpto
> isolde jumpto
> isolde jumpto
> select clear
> isolde jumpto
> select clear
> isolde jumpto
> isolde jumpto
> select clear
> aw
> isolde sim start sel
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> select clear
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
Traceback (most recent call last):
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2708, in _start_sim_or_toggle_pause
self.start_sim()
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2725, in start_sim
main_sel = self._last_main_sel = self._get_main_sim_selection()
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2908, in _get_main_sim_selection
raise TypeError('You must select at least one atom from the current '
TypeError: You must select at least one atom from the current working model
prior to starting a simulation!
TypeError: You must select at least one atom from the current working model
prior to starting a simulation!
File "/home/tic20/.local/share/ChimeraX/1.1/site-
packages/chimerax/isolde/isolde.py", line 2908, in _get_main_sim_selection
raise TypeError('You must select at least one atom from the current '
See log for complete Python traceback.
> isolde jumpto
> select clear
> aw
> isolde sim start sel
> isolde jumpto
> isolde jumpto
> select clear
> isolde jumpto
> select clear
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> select clear
> isolde jumpto
> isolde jumpto
> isolde jumpto
> isolde jumpto
> save working_fme.cxs
Taking snapshot of stepper: Model 1a
> select /A*,a*:1
800 atoms, 760 bonds, 40 residues, 1 model selected
> select clear
> save working_fme.cxs
Taking snapshot of stepper: Model 1a
opened ChimeraX session
> isolde start
> set selectionWidth 4
Done loading forcefield
> select clear
> ui tool show Shell
/opt/UCSF/ChimeraX/lib/python3.7/site-packages/IPython/core/history.py:226:
UserWarning: IPython History requires SQLite, your history will not be saved
warn("IPython History requires SQLite, your history will not be saved")
> select ~protein&~:HOH
34491 atoms, 35465 bonds, 14 pseudobonds, 320 residues, 21 models selected
> select clear
> aw
> isolde sim start sel
> select clear
> select clear
> aw
> isolde sim start sel
> delete sel
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select up
138 atoms, 139 bonds, 1 residue, 1 model selected
> show sel
> awsf
> awsf
> aw
> isolde sim start sel
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select up
140 atoms, 148 bonds, 1 residue, 1 model selected
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> select ~protein&~:HOH
34491 atoms, 35465 bonds, 14 pseudobonds, 320 residues, 22 models selected
> clipper isolate sel contextDistance 0
> hide #!8.1 models
> ~cartoon
> cartoon
> show #!8.1 models
> save working.cxs
Taking snapshot of stepper: Model 1a
> isolde add ligand MQ9
> ui mousemode right "translate selected atoms"
> select up
138 atoms, 139 bonds, 1 residue, 1 model selected
> isolde ignore ~sel
ISOLDE: currently ignoring 5734 residues in model 8.2
> isolde ~ignore
> select up
138 atoms, 139 bonds, 1 residue, 1 model selected
> select clear
> select up
138 atoms, 139 bonds, 1 residue, 1 model selected
> delete sel
> select clear
> select clear
> select clear
> select clear
> select clear
> select clear
> awsf
> awsf
> select clear
> select clear
> select clear
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> select clear
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> select clear
> select clear
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> select clear
> aw
> isolde sim start sel
> select clear
> select clear
> select clear
> clipper set contourSensitivity 0.25
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> select clear
> select clear
> aw
> isolde sim start sel
> aw
> isolde sim start sel
> awsf
> awsf
> aw
> isolde sim start sel
> select clear
> aw
> isolde sim start sel
> select clear
> aw
> isolde sim start sel
> select clear
> aw
> isolde sim start sel
> select clear
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> awsf
> awsf
> awsf
> select clear
> select up
6 atoms, 4 bonds, 2 residues, 1 model selected
> delete sel
> select clear
> save working.cxs
Taking snapshot of stepper: Model 1a
> select clear
> save working.cxs
Taking snapshot of stepper: Model 1a
> save working.cxs
Taking snapshot of stepper: Model 1a
> open ../jsg.pdb
Summary of feedback from opening ../jsg.pdb
---
warnings | Start residue of secondary structure not found: HELIX 1 1 THR A 43
GLY A 51 1 9
Start residue of secondary structure not found: HELIX 2 2 PRO A 71 ALA A 76 1
6
Start residue of secondary structure not found: HELIX 3 3 SER A 94 VAL A 102 1
9
Start residue of secondary structure not found: HELIX 4 4 ARG A 114 PHE A 125
1 12
Start residue of secondary structure not found: HELIX 5 5 ARG A 144 LEU A 152
1 9
60 messages similar to the above omitted
> select #1
200 atoms, 206 bonds, 1 residue, 1 model selected
> close #1
> open ../jsg.pdb
Summary of feedback from opening ../jsg.pdb
---
warnings | Start residue of secondary structure not found: HELIX 1 1 THR A 43
GLY A 51 1 9
Start residue of secondary structure not found: HELIX 2 2 PRO A 71 ALA A 76 1
6
Start residue of secondary structure not found: HELIX 3 3 SER A 94 VAL A 102 1
9
Start residue of secondary structure not found: HELIX 4 4 ARG A 114 PHE A 125
1 12
Start residue of secondary structure not found: HELIX 5 5 ARG A 144 LEU A 152
1 9
60 messages similar to the above omitted
> select #1
200 atoms, 206 bonds, 1 residue, 1 model selected
> ui mousemode right "translate selected atoms"
> close #1
> select clear
> select clear
> select clear
> isolde add ligand GAL
Deleted the following atoms from residue GAL L837: O1, HO1
> show sel
> ui mousemode right "translate selected atoms"
> delete sel
> isolde add ligand GLC
Deleted the following atoms from residue GLC L837: HO1, O1
> show sel
> delete sel
> isolde add ligand NAG
Deleted the following atoms from residue NAG L837: O1, HO1
> show sel
> delete sel
> select clear
> aw
> isolde sim start sel
> select up
133 atoms, 132 bonds, 1 residue, 1 model selected
> select clear
> select up
141 atoms, 141 bonds, 1 residue, 1 model selected
> select up
141 atoms, 141 bonds, 1 residue, 1 model selected
> awsf
> awsf
> ui mousemode right "mark point"
> marker #1 position 206,174.5,206.2 color yellow radius 1
> marker #1 position 208.1,175.1,203.3 color yellow radius 1
> marker #1 position 210.5,177.7,202.2 color yellow radius 1
> marker #1 position 209.7,176.4,199 color yellow radius 1
> marker #1 position 213.2,175.9,196.8 color yellow radius 1
> delete sel
> marker #1 position 211.3,175.1,196.4 color yellow radius 1
> marker #1 position 214.6,172.1,194.3 color yellow radius 1
> delete sel
> marker #1 position 213.4,173.4,193.2 color yellow radius 1
> marker #1 position 216.9,174.5,192.6 color yellow radius 1
> delete sel
> marker #1 position 215.5,174.8,190.6 color yellow radius 1
> marker #1 position 214.8,175.9,188.4 color yellow radius 1
> view /aj:12
> marker #1 position 198.9,164,186.4 color yellow radius 1
> marker #1 position 199.1,162.2,189.3 color yellow radius 1
> marker #1 position 197.6,159.5,191.3 color yellow radius 1
> marker #1 position 197.1,156.2,191.5 color yellow radius 1
> marker #1 position 196.1,153.9,193.2 color yellow radius 1
> marker #1 position 195,153.8,196.9 color yellow radius 1
> marker #1 position 195.7,152.3,200.4 color yellow radius 1
> marker #1 position 195.6,150.6,201.3 color yellow radius 1
> marker #1 position 193.5,148.1,198.7 color yellow radius 1
> marker #1 position 194.5,149.2,200.1 color yellow radius 1
> marker #1 position 193.2,148,196.4 color yellow radius 1
> marker #1 position 197.7,166.8,183.9 color yellow radius 1
> select /a*:12
384 atoms, 368 bonds, 16 residues, 1 model selected
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2021_01_final_maps/model1a/class4b_r_2.35A_postprocess_masked.mrc
Opened class4b_r_2.35A_postprocess_masked.mrc, grid size 400,400,400, pixel
0.999, shown at level 0.0164, step 2, values float32
> volume #8.1.1.1 level 0.03142
> volume #8.1.1.1 level 0.02833
> volume zone #2 nearAtoms #1 range 5
> volume #2 step 1
> volume #2 level 0.03435
> volume #2 level 0.02539
> volume zone #2 nearAtoms #1 range 4
> volume #2 level 0.0224
> save unknown_density_tubes.mrc #2
> usage volume zone
volume zone volumes nearAtoms an atoms specifier [range a number]
[bondPointSpacing a number] [minimalBounds true or false] [newMap true or
false] [invert true or false] [subregion map region] [step map step] [modelId
modelId]
— Zero map values beyond a distance range from atoms
modelId: a model id
> volume zone #2 nearAtoms #1 range 4 minimalBounds true
> select #2
4 models selected
> volume sel showOutlineBox true
> save unknown_density_tubes.mrc #2
> open unknown_density_tubes.mrc
Opened unknown_density_tubes.mrc, grid size 32,39,32, pixel 0.999,0.999,0.999,
shown at level 0.0972, step 1, values float32
> hide #!2 models
> volume #3 level 0.05529
> volume #3 level 0.05219
> volume #3 level 0.02932
> volume #3 level 0.04173
> close #3
> show #!2 models
> save unknown_density_tubes.ccp4 #2
No known data format for file suffix '.ccp4'
> save unknown_density_tubes.map #2
No known data format for file suffix '.map'
> surface unzone #2
> volume zone #2 nearAtoms #1 range 4
> surface unzone #2
> volume zone #2 nearAtoms #1 range 4 minimalBounds true newMap true
> hide #!3 models
> show #!3 models
> surface unzone #3
> save unknown_density_tubes.mrc #3
> open unknown_density_tubes.mrc
Opened unknown_density_tubes.mrc, grid size 32,39,32, pixel 0.999,0.999,0.999,
shown at level 0.0254, step 1, values float32
> hide #!3 models
> close #1#2-4
> select clear
> select clear
> aw
> isolde sim start sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> select clear
> aw
> isolde sim start sel
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> aw
> isolde sim start sel
> select clear
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> delete sel
> select clear
> select clear
> select clear
> select clear
> save working.cxs
Taking snapshot of stepper: Model 1a
Restoring stepper: Model 1a
opened ChimeraX session
> volume unzone
Missing or invalid "volumes" argument: empty atom specifier
> volume unzone #8
> close #8.1
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2021_01_final_maps/model1a/class4b_r_2.35A_postprocess_masked.mrc
Opened class4b_r_2.35A_postprocess_masked.mrc, grid size 400,400,400, pixel
0.999, shown at level 0.0164, step 2, values float32
> volume gaussian #1 bfactor 50
> clipper associate #1,2 toModel #8
> clipper set contourSensitivity 0.25
> select ~protein&~:HOH
34491 atoms, 35465 bonds, 14 pseudobonds, 320 residues, 21 models selected
> select clear
> select clear
> save working.cxs
Taking snapshot of stepper: Model 1a
> save working.cxs
Taking snapshot of stepper: Model 1a
> select clear
> select up
124 atoms, 122 bonds, 2 residues, 1 model selected
> delete sel
> isolde add ligand CDL
place_ligand() was called with use_md_template=True, but no suitable template
was found. This command has been ignored.
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> ui mousemode right "translate selected atoms"
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> isolde ignore ~sel
ISOLDE: currently ignoring 5754 residues in model 8.2
> isolde ~ignore
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> select clear
> select up
256 atoms, 255 bonds, 1 residue, 1 model selected
> select clear
> isolde ~ignore
> select clear
> select clear
> select clear
> select up
49 atoms, 50 bonds, 2 residues, 1 model selected
> select clear
> save working.cxs
Taking snapshot of stepper: Model 1a
> select #1
Nothing selected
> select #8
114704 atoms, 116622 bonds, 15 pseudobonds, 5755 residues, 23 models selected
> select clear
> volume zone #8.1.1.1 nearAtoms #8 range 2.5 newMap true
> volume subtract #8.1.1.1 #1
> close #1
> show #!8.1.1.1 models
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> isolde ignore ~sel
ISOLDE: currently ignoring 5755 residues in model 8.2
> isolde ~ignore
> hide #!2 models
> ui mousemode right "mark point"
> marker #1 position 243.7,161,187.6 color yellow radius 1
> ui mousemode right "resize markers"
> marker change #1:24 radius 3.127
> show #!2 models
> hide #!2 models
> isolde add ligand LMT
> ui mousemode right "translate selected atoms"
> select up
14 atoms, 13 bonds, 1 residue, 1 model selected
> select up
50 atoms, 49 bonds, 4 residues, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 4 residues in model 8.2
> isolde ~ignore
> select clear
> select up
81 atoms, 82 bonds, 1 residue, 1 model selected
> delete sel
> ui mousemode right "mark point"
> marker #1 position 204.4,140.7,186.9 color yellow radius 3.127
> color sel red
> select clear
> show #!2 models
> hide #!2 models
> show #!2 models
> hide #1 models
> sequence chain /C
Chains must have same sequence
> sequence chain #8/C
Chains must have same sequence
> select #8/C
5225 atoms, 5244 bonds, 4 pseudobonds, 395 residues, 2 models selected
> hide #!2 models
> select clear
> select clear
> delete sel
> save working.cxs
Taking snapshot of stepper: Model 1a
> select up
216 atoms, 217 bonds, 12 residues, 1 model selected
> select clear
> select clear
> save working.cxs
Taking snapshot of stepper: Model 1a
> save model1a_fully_rebuilt.cif #8
Not saving entity_poly_seq for non-authoritative sequences
> save working.cxs
Taking snapshot of stepper: Model 1a
opened ChimeraX session
> select #1/A*:BCL
Nothing selected
> select #8/A*:BCL
6720 atoms, 7104 bonds, 48 residues, 1 model selected
> select clear
> isolde start
> set selectionWidth 4
Done loading forcefield
> select #8/A*:BCL
6720 atoms, 7104 bonds, 48 residues, 1 model selected
> select clear
> awsf
> awsf
> awsf
> awsf
> open
> /run/media/tic20/storage/structure_dump/collaboration/pu_qian/2021_01_final_maps/model1a/model1a_fully_rebuilt.cif
Summary of feedback from opening
/run/media/tic20/storage/structure_dump/collaboration/pu_qian/2021_01_final_maps/model1a/model1a_fully_rebuilt.cif
---
warnings | Unknown polymer entity '1' near line 610
Unknown polymer entity '2' near line 2941
Unknown polymer entity '3' near line 6564
Unknown polymer entity '4' near line 29734
Unknown polymer entity '5' near line 30743
5 messages similar to the above omitted
Unable to fetch template for 'LMX': might have incorrect bonds
Unknown polymer entity '11' near line 71410
Unknown polymer entity '12' near line 78114
Unknown polymer entity '13' near line 89914
Unknown polymer entity '14' near line 95896
Atom C1 is not in the residue template for GPC /AA:201
Atom C1 is not in the residue template for GPC /AB:201
Atom C1 is not in the residue template for GPC /AC:201
Atom C1 is not in the residue template for GPC /AD:201
Atom C1 is not in the residue template for GPC /AE:201
19 messages similar to the above omitted
Atom H is not in the residue template for GLY /BQ:5
Atom H is not in the residue template for GLY /BR:5
Atom H is not in the residue template for GLY /BS:5
Atom H is not in the residue template for GLY /BT:5
Atom H is not in the residue template for GLY /C:13
Atom O2 is not in the residue template for GUX /C:1001
Atom H6 is not in the residue template for P5S /H1:1001
Atom H11 is not in the residue template for BPH /L:606
Atom H11 is not in the residue template for BPH /M:605
Atom C1 is not in the residue template for RCC /M:701
Atom O2 is not in the residue template for GUX /M:1001
Atom HN2 is not in the residue template for ARG /ak:71
Atom C1 is not in the residue template for GPC /ba:100
Atom C1 is not in the residue template for GPC /bb:100
Atom C26 is not in the residue template for PEX /bb:203
Atom C26 is not in the residue template for PEX /bb:204
Atom C1 is not in the residue template for GPC /bc:100
Atom C1 is not in the residue template for GPC /bd:100
Atom C26 is not in the residue template for PEX /bd:211
Atom C26 is not in the residue template for PEX /bd:209
Atom C1 is not in the residue template for GPC /be:100
Atom C26 is not in the residue template for PEX /be:205
Atom C1 is not in the residue template for GPC /bf:100
Atom C26 is not in the residue template for PEX /bf:212
Atom C1 is not in the residue template for GPC /bg:100
Atom C1 is not in the residue template for GPC /bh:100
Atom C26 is not in the residue template for PEX /bh:209
Atom C26 is not in the residue template for PEX /bh:207
Atom C1 is not in the residue template for GPC /bi:100
Atom C1 is not in the residue template for GPC /bj:100
Atom C26 is not in the residue template for PEX /bj:210
Atom C26 is not in the residue template for PEX /bj:208
Atom C1 is not in the residue template for GPC /bk:100
Atom C1 is not in the residue template for GPC /bl:100
Atom C26 is not in the residue template for PEX /bl:204
Atom C26 is not in the residue template for PEX /bl:206
Atom C1 is not in the residue template for GPC /bm:100
Atom C1 is not in the residue template for GPC /bn:100
Atom C26 is not in the residue template for PEX /bn:209
Atom C26 is not in the residue template for PEX /bn:207
Atom C1 is not in the residue template for GPC /bo:100
Atom C26 is not in the residue template for PEX /bp:206
Atom C1 is not in the residue template for GPC /bp:100
Atom C26 is not in the residue template for PEX /bp:204
Atom O2 is not in the residue template for GUX /M:1001
Atom O2 is not in the residue template for GUX /C:1001
Missing or incomplete entity_poly_seq table. Inferred polymer connectivity.
Chain information for model1a_fully_rebuilt.cif #3
---
Chain | Description
AA AB AE AF AG AH AI AJ AK AL AM AN AO AP AQ AR AS AT AU AV AW AX | ?
AC AD | ?
BA BC BF BG BH BJ BK BL BM BN BO BP BU BX ba bb bc bd be bf bg bh bi bj bk bl
bm bo bp | ?
BB BD BE BI BQ BR BS BT BV BW bn | ?
C | ?
C1 | ?
H1 | ?
H2 | ?
L | ?
M | ?
aa ab ad ae af ag ah ai aj al am ao | ?
ac | ?
ak an | ?
ap | ?
> hide #1*protein
Expected a collection of one of 'atoms', 'bonds', 'cartoons', 'models',
'pbonds', 'pseudobonds', 'ribbons', or 'surfaces' or a keyword
> hide #1&protein
> hide #3&protein
> close #1#2
> matchmaker #3/AC to #8/AB
Parameters
---
Chain pairing | bb
Alignment algorithm | Needleman-Wunsch
Similarity matrix | BLOSUM-62
SS fraction | 0.3
Gap open (HH/SS/other) | 18/18/6
Gap extend | 1
SS matrix | | | H | S | O
---|---|---|---
H | 6 | -9 | -6
S | | 6 | -6
O | | | 4
Iteration cutoff | 2
Matchmaker Model 1a, chain AB (#8.2) with model1a_fully_rebuilt.cif, chain AC
(#3), sequence alignment score = 261.6
RMSD between 48 pruned atom pairs is 0.331 angstroms; (across all 48 pairs:
0.331)
> color #3 tan
> color byhetero
> hide #!3 models
> awsf
> awsf
> select clear
> show #!3 models
> matchmaker #3/AG to #8/AF
Parameters
---
Chain pairing | bb
Alignment algorithm | Needleman-Wunsch
Similarity matrix | BLOSUM-62
SS fraction | 0.3
Gap open (HH/SS/other) | 18/18/6
Gap extend | 1
SS matrix | | | H | S | O
---|---|---|---
H | 6 | -9 | -6
S | | 6 | -6
O | | | 4
Iteration cutoff | 2
Matchmaker Model 1a, chain AF (#8.2) with model1a_fully_rebuilt.cif, chain AG
(#3), sequence alignment score = 262.1
RMSD between 49 pruned atom pairs is 0.317 angstroms; (across all 49 pairs:
0.317)
> hide #!3 models
> select clear
> aw
> isolde sim start sel
> awsf
> awsf
> awsf
> awsf
> show #!3 models
> matchmaker #3/AM to #1/AL
No 'to' model specified
> hide #!3 models
> awsf
> awsf
> awsf
> awsf
> show #!3 models
> matchmaker #3/AP to #1/AO
No 'to' model specified
> matchmaker #3/AP to #8/AO
Parameters
---
Chain pairing | bb
Alignment algorithm | Needleman-Wunsch
Similarity matrix | BLOSUM-62
SS fraction | 0.3
Gap open (HH/SS/other) | 18/18/6
Gap extend | 1
SS matrix | | | H | S | O
---|---|---|---
H | 6 | -9 | -6
S | | 6 | -6
O | | | 4
Iteration cutoff | 2
Matchmaker Model 1a, chain AO (#8.2) with model1a_fully_rebuilt.cif, chain AP
(#3), sequence alignment score = 271.3
RMSD between 49 pruned atom pairs is 0.357 angstroms; (across all 49 pairs:
0.357)
> hide #!3 models
> awsf
> awsf
> awsf
> select clear
> show #!3 models
> hide #!3 models
> awsf
> awsf
> select clear
> awsf
> awsf
> show #!3 models
> hide #!3 models
> awsf
> awsf
> awsf
> awsf
> select clear
> save working.cxs
Taking snapshot of stepper: Model 1a
> select #8/A*:BCL
6720 atoms, 7104 bonds, 48 residues, 1 model selected
> select clear
> select #8/A*:BCL
6720 atoms, 7104 bonds, 48 residues, 1 model selected
> save working.cxs
Taking snapshot of stepper: Model 1a
opened ChimeraX session
> isolde start
> set selectionWidth 4
Done loading forcefield
> view /M:609
No objects specified.
> view /M:608
> select clear
> select clear
> select clear
> close #1
> select up
138 atoms, 139 bonds, 1 residue, 1 model selected
> isolde replace ligand sel MQ8
Deleted the following atoms from residue MQ9 M608: H48, C47, H512, C49, H501,
C50, H471, H503, H511, C48, H513, H502, C51, H472
124 atoms were automatically renamed to match the template: O1->O4, C1->C4,
C2->C5, C6->C3, C7->C11, C5->C2, C8->C12, C9->C13, H8->H121, C10->C14,
C11->C15, C12->C16, C13->C17, C14->C18, H13->H171, C15->C19, C16->C20,
C17->C21, C18->C22, C19->C23, H18->H221, C20->C24, C21->C25, C22->C26,
C23->C27, C24->C28, H23->H271, C25->C29, C26->C30, C27->C31, C28->C32,
C29->C33, H28->H321, C30->C34, C31->C35, C32->C36, C33->C37, C34->C38,
H33->H371, C35->C39, C36->C40, C37->C41, C38->C42, C39->C43, H38->H421,
C40->C45, C41->C44, C42->C46, C43->C47, C44->C48, H43->H471, C45->C49,
C46->C50, C5M->C2M, C4->C1, C3->C10, C3A->C9, C3B->C8, C3C->C7, C3D->C6,
O4->O1, H3A->H91, H3B->H81, H3C->H71, H3D->H61, H111->H151, H112->H152,
H121->H161, H122->H162, H161->H201, H162->H202, H171->H211, H172->H212,
H211->H251, H212->H252, H221->H261, H222->H262, H261->H301, H262->H302,
H271->H311, H272->H312, H311->H351, H312->H352, H321->H361, H322->H362,
H361->H401, H362->H402, H371->H411, H372->H412, H411->H441, H412->H442,
H421->H461, H422->H462, H71->H111, H72->H112, H461->H501, H462->H502,
H101->H141, H102->H142, H103->H143, H151->H191, H152->H192, H153->H193,
H201->H241, H202->H242, H203->H243, H251->H291, H252->H292, H253->H293,
H301->H341, H302->H342, H303->H343, H351->H391, H352->H392, H353->H393,
H401->H451, H402->H452, H403->H453, H451->H491, H452->H492, H453->H493,
H5M1->H2M1, H5M2->H2M2, H5M3->H2M3
> show sel
> select :CDL
1536 atoms, 1530 bonds, 6 residues, 1 model selected
> select clear
> save working.cxs
Taking snapshot of stepper: Model 1a
> view /M:608
> select up
125 atoms, 126 bonds, 1 residue, 1 model selected
> save working.cxs
Taking snapshot of stepper: Model 1a
> view /M:701
> select /M:701
100 atoms, 99 bonds, 1 residue, 1 model selected
> movie record
> movie stop
> movie encode RCC.mp4
Movie saved to RCC.mp4
> windowsize size
Expected an integer >= 1 or a keyword
> windowsize
window size 1136 934
> windowsize
window size 1136 934
> show sel
> select clear
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
Tugging of non-polar hydrogens is not enabled. Applying tug to the nearest
bonded heavy atom.
> isolde replace ligand sel CRT
Timed out trying to match residue RCC to template CRT. Match may not be ideal.
Deleted the following atoms from residue RCC M701: H27B, C27, O45, H24B, H43C,
C24, O42, H38A, H23A, C39, H28B, C29, C26, O44, H38C, H27A, H24A, C23, H43B,
C38, H28A, C28, HC25, C25, C43, H38B, H23B, C40, HC29, H43A
68 atoms were automatically renamed to match the template: O32->O1, C41->C1M,
C2->C4, C3->C5, C4->C6, C5->C7, HC3->H5, HC4->H6, C30->C2, C31->C3, C33->C8,
C6->C9, C7->C10, C8->C11, C9->C12, HC6->H9, HC7->H10, HC8->H11, C10->C14,
C11->C15, C12->C16, C13->C17, C34->C13, HC10->H14, HC11->H15, HC12->H16,
C35->C18, C14->C19, C15->C20, C16->C21, C17->C22, C18->C23, HC14->H19,
HC15->H20, HC16->H21, HC17->H22, C36->C24, C19->C25, C20->C26, C21->C27,
C22->C28, HC19->H25, HC20->H26, HC21->H27, C37->C29, HC2A->H41, HC2B->H42,
H30A->H21A, H30B->H22A, H30C->H23, H31B->H32A, H31C->H33, H33A->H81,
H33B->H82, H33C->H83, HC34->H131, H35A->H181, H35B->H182, H35C->H183,
H36A->H241, H36B->H242, H36C->H243, H37A->H291, H37B->H292, H37C->H293,
H41A->H1M1, H41B->H1M2, H41C->H1M3
> select up
153 atoms, 152 bonds, 2 residues, 1 model selected
> isolde ignore sel
ISOLDE: currently ignoring 2 residues in model 8.2
> isolde ~ignore
> save working.cxs
Taking snapshot of stepper: Model 1a
> select up
3 atoms, 2 bonds, 1 residue, 1 model selected
> delete sel
> save working.cxs
Taking snapshot of stepper: Model 1a
> view /ah:11
> select /ag:12
24 atoms, 23 bonds, 1 residue, 1 model selected
> select /ah,ag:12
48 atoms, 46 bonds, 2 residues, 1 model selected
> view /M:36
> ui tool show Shell
/opt/UCSF/ChimeraX/lib/python3.7/site-packages/IPython/core/history.py:226:
UserWarning: IPython History requires SQLite, your history will not be saved
warn("IPython History requires SQLite, your history will not be saved")
> save model_1a_working.cif #1
no structures to save
> save working.cxs
Taking snapshot of stepper: Model 1a
opened ChimeraX session
> ui tool show Shell
/opt/UCSF/ChimeraX/lib/python3.7/site-packages/IPython/core/history.py:226:
UserWarning: IPython History requires SQLite, your history will not be saved
warn("IPython History requires SQLite, your history will not be saved")
> select /M:MAN
20 atoms, 20 bonds, 1 residue, 1 model selected
> view sel
> select up
49 atoms, 50 bonds, 2 residues, 1 model selected
> hide #!8.1 models
> set bgColor black
> hide :HOH
> select up
59 atoms, 60 bonds, 3 residues, 1 model selected
> hide protein&~sel
> select /M:1000@O5
1 atom, 1 residue, 1 model selected
> select up
3 atoms, 1 bond, 2 residues, 1 model selected
> select up
49 atoms, 50 bonds, 2 residues, 1 model selected
> save duplicated_sugars.jpg
OpenGL version: 3.3.0 NVIDIA 455.32.00
OpenGL renderer: TITAN Xp/PCIe/SSE2
OpenGL vendor: NVIDIA Corporation
Manufacturer: Dell Inc.
Model: Precision T5600
OS: CentOS Linux 7 Core
Architecture: 64bit ELF
CPU: 32 Intel(R) Xeon(R) CPU E5-2687W 0 @ 3.10GHz
Cache Size: 20480 KB
Memory:
total used free shared buff/cache available
Mem: 62G 8.7G 40G 311M 13G 53G
Swap: 4.9G 0B 4.9G
Graphics:
03:00.0 VGA compatible controller [0300]: NVIDIA Corporation GP102 [TITAN Xp] [10de:1b02] (rev a1)
Subsystem: NVIDIA Corporation Device [10de:11df]
Kernel driver in use: nvidia
PyQt version: 5.12.3
Compiled Qt version: 5.12.4
Runtime Qt version: 5.12.9
Installed Packages:
alabaster: 0.7.12
appdirs: 1.4.4
Babel: 2.8.0
backcall: 0.2.0
blockdiag: 2.0.1
certifi: 2020.6.20
chardet: 3.0.4
ChimeraX-AddH: 2.1.1
ChimeraX-AlignmentAlgorithms: 2.0
ChimeraX-AlignmentHdrs: 3.2
ChimeraX-AlignmentMatrices: 2.0
ChimeraX-Alignments: 2.1
ChimeraX-Arrays: 1.0
ChimeraX-Async: 0.1
ChimeraX-Atomic: 1.6.1
ChimeraX-AtomSearch: 2.0
ChimeraX-AxesPlanes: 2.0
ChimeraX-BasicActions: 1.1
ChimeraX-BILD: 1.0
ChimeraX-BlastProtein: 1.0.1
ChimeraX-BondRot: 2.0
ChimeraX-BugReporter: 1.0
ChimeraX-BuildStructure: 2.0
ChimeraX-Bumps: 1.0
ChimeraX-BundleBuilder: 1.0
ChimeraX-ButtonPanel: 1.0
ChimeraX-CageBuilder: 1.0
ChimeraX-CellPack: 1.0
ChimeraX-Centroids: 1.1
ChimeraX-ChemGroup: 2.0
ChimeraX-Clashes: 2.0
ChimeraX-Clipper: 0.15.0
ChimeraX-ColorActions: 1.0
ChimeraX-ColorGlobe: 1.0
ChimeraX-CommandLine: 1.1.3
ChimeraX-ConnectStructure: 2.0
ChimeraX-Contacts: 1.0
ChimeraX-Core: 1.1
ChimeraX-CoreFormats: 1.0
ChimeraX-coulombic: 1.0.1
ChimeraX-Crosslinks: 1.0
ChimeraX-Crystal: 1.0
ChimeraX-DataFormats: 1.0
ChimeraX-Dicom: 1.0
ChimeraX-DistMonitor: 1.1
ChimeraX-DistUI: 1.0
ChimeraX-Dssp: 2.0
ChimeraX-EMDB-SFF: 1.0
ChimeraX-ExperimentalCommands: 1.0
ChimeraX-FileHistory: 1.0
ChimeraX-FunctionKey: 1.0
ChimeraX-Geometry: 1.1
ChimeraX-gltf: 1.0
ChimeraX-Graphics: 1.0
ChimeraX-Hbonds: 2.0
ChimeraX-Help: 1.0
ChimeraX-HKCage: 1.3
ChimeraX-IHM: 1.0
ChimeraX-ImageFormats: 1.0
ChimeraX-IMOD: 1.0
ChimeraX-IO: 1.0
ChimeraX-ISOLDE: 1.1.0
ChimeraX-Label: 1.0
ChimeraX-LinuxSupport: 1.0
ChimeraX-ListInfo: 1.0
ChimeraX-Log: 1.1.1
ChimeraX-LookingGlass: 1.1
ChimeraX-Map: 1.0.1
ChimeraX-MapData: 2.0
ChimeraX-MapEraser: 1.0
ChimeraX-MapFilter: 2.0
ChimeraX-MapFit: 2.0
ChimeraX-MapSeries: 2.0
ChimeraX-Markers: 1.0
ChimeraX-Mask: 1.0
ChimeraX-MatchMaker: 1.1
ChimeraX-MDcrds: 2.0
ChimeraX-MedicalToolbar: 1.0.1
ChimeraX-Meeting: 1.0
ChimeraX-MLP: 1.0
ChimeraX-mmCIF: 2.2
ChimeraX-MMTF: 2.0
ChimeraX-Modeller: 1.0
ChimeraX-ModelPanel: 1.0
ChimeraX-ModelSeries: 1.0
ChimeraX-Mol2: 2.0
ChimeraX-Morph: 1.0
ChimeraX-MouseModes: 1.0
ChimeraX-Movie: 1.0
ChimeraX-Neuron: 1.0
ChimeraX-Nucleotides: 2.0
ChimeraX-OpenCommand: 1.2.1
ChimeraX-PDB: 2.1
ChimeraX-PDBBio: 1.0
ChimeraX-Phenix: 0.1
ChimeraX-PickBlobs: 1.0
ChimeraX-Positions: 1.0
ChimeraX-PresetMgr: 1.0
ChimeraX-PubChem: 2.0
ChimeraX-Read-Pbonds: 1.0
ChimeraX-Registration: 1.1
ChimeraX-RemoteControl: 1.0
ChimeraX-ResidueFit: 1.0
ChimeraX-RestServer: 1.0
ChimeraX-RNALayout: 1.0
ChimeraX-RotamerLibMgr: 2.0
ChimeraX-RotamerLibsDunbrack: 2.0
ChimeraX-RotamerLibsDynameomics: 2.0
ChimeraX-RotamerLibsRichardson: 2.0
ChimeraX-SaveCommand: 1.2
ChimeraX-SchemeMgr: 1.0
ChimeraX-SDF: 2.0
ChimeraX-Segger: 1.0
ChimeraX-Segment: 1.0
ChimeraX-SeqView: 2.2
ChimeraX-Shape: 1.0.1
ChimeraX-Shell: 1.0
ChimeraX-Shortcuts: 1.0
ChimeraX-ShowAttr: 1.0
ChimeraX-ShowSequences: 1.0
ChimeraX-SideView: 1.0
ChimeraX-Smiles: 2.0
ChimeraX-SmoothLines: 1.0
ChimeraX-SpaceNavigator: 1.0
ChimeraX-StdCommands: 1.0.4
ChimeraX-STL: 1.0
ChimeraX-Storm: 1.0
ChimeraX-Struts: 1.0
ChimeraX-Surface: 1.0
ChimeraX-SwapAA: 2.0
ChimeraX-SwapRes: 2.0
ChimeraX-TapeMeasure: 1.0
ChimeraX-Test: 1.0
ChimeraX-Toolbar: 1.0
ChimeraX-ToolshedUtils: 1.0
ChimeraX-Tug: 1.0
ChimeraX-UI: 1.2.3
ChimeraX-uniprot: 2.0
ChimeraX-ViewDockX: 1.0
ChimeraX-Vive: 1.1
ChimeraX-VolumeMenu: 1.0
ChimeraX-VTK: 1.0
ChimeraX-WavefrontOBJ: 1.0
ChimeraX-WebCam: 1.0
ChimeraX-WebServices: 1.0
ChimeraX-Zone: 1.0
colorama: 0.4.3
comtypes: 1.1.7
cxservices: 1.0
cycler: 0.10.0
Cython: 0.29.20
decorator: 4.4.2
distlib: 0.3.1
distro: 1.5.0
docutils: 0.16
filelock: 3.0.12
funcparserlib: 0.3.6
grako: 3.16.5
graphviz: 0.14.1
h5py: 2.10.0
html2text: 2020.1.16
idna: 2.10
ihm: 0.16
imagecodecs: 2020.5.30
imagecodecs-lite: 2020.1.31
imagesize: 1.2.0
ipykernel: 5.3.0
ipython: 7.15.0
ipython-genutils: 0.2.0
jedi: 0.17.2
Jinja2: 2.11.2
jupyter-client: 6.1.3
jupyter-core: 4.6.3
kiwisolver: 1.2.0
line-profiler: 2.1.2
lxml: 4.5.1
MarkupSafe: 1.1.1
matplotlib: 3.2.1
msgpack: 1.0.0
netifaces: 0.10.9
networkx: 2.4
numexpr: 2.7.1
numpy: 1.18.5
numpydoc: 1.0.0
objgraph: 3.4.1
openvr: 1.12.501
packaging: 20.4
ParmEd: 3.2.0
parso: 0.7.1
pexpect: 4.8.0
pickleshare: 0.7.5
Pillow: 7.1.2
pip: 20.2.2
pkginfo: 1.5.0.1
prompt-toolkit: 3.0.7
psutil: 5.7.0
ptyprocess: 0.6.0
pycollada: 0.7.1
pydicom: 2.0.0
Pygments: 2.6.1
PyOpenGL: 3.1.5
PyOpenGL-accelerate: 3.1.5
pyparsing: 2.4.7
PyQt5-commercial: 5.12.3
PyQt5-sip: 4.19.19
PyQtWebEngine-commercial: 5.12.1
python-dateutil: 2.8.1
pytz: 2020.1
pyzmq: 19.0.2
qtconsole: 4.7.4
QtPy: 1.9.0
RandomWords: 0.3.0
requests: 2.24.0
scipy: 1.4.1
Send2Trash: 1.5.0
SEQCROW: 0.20
setuptools: 49.4.0
sfftk-rw: 0.6.6.dev0
six: 1.15.0
snowballstemmer: 2.0.0
sortedcontainers: 2.2.2
Sphinx: 3.1.1
sphinxcontrib-applehelp: 1.0.2
sphinxcontrib-blockdiag: 2.0.0
sphinxcontrib-devhelp: 1.0.2
sphinxcontrib-htmlhelp: 1.0.3
sphinxcontrib-jsmath: 1.0.1
sphinxcontrib-qthelp: 1.0.3
sphinxcontrib-serializinghtml: 1.1.4
suds-jurko: 0.6
tables: 3.6.1
tifffile: 2020.6.3
tinyarray: 1.2.2
tornado: 6.0.4
traitlets: 5.0.4
urllib3: 1.25.10
versioneer: 0.18
wcwidth: 0.2.5
webcolors: 1.11.1
wheel: 0.34.2
File attachment: duplicated_sugars.jpg
{kind=link}
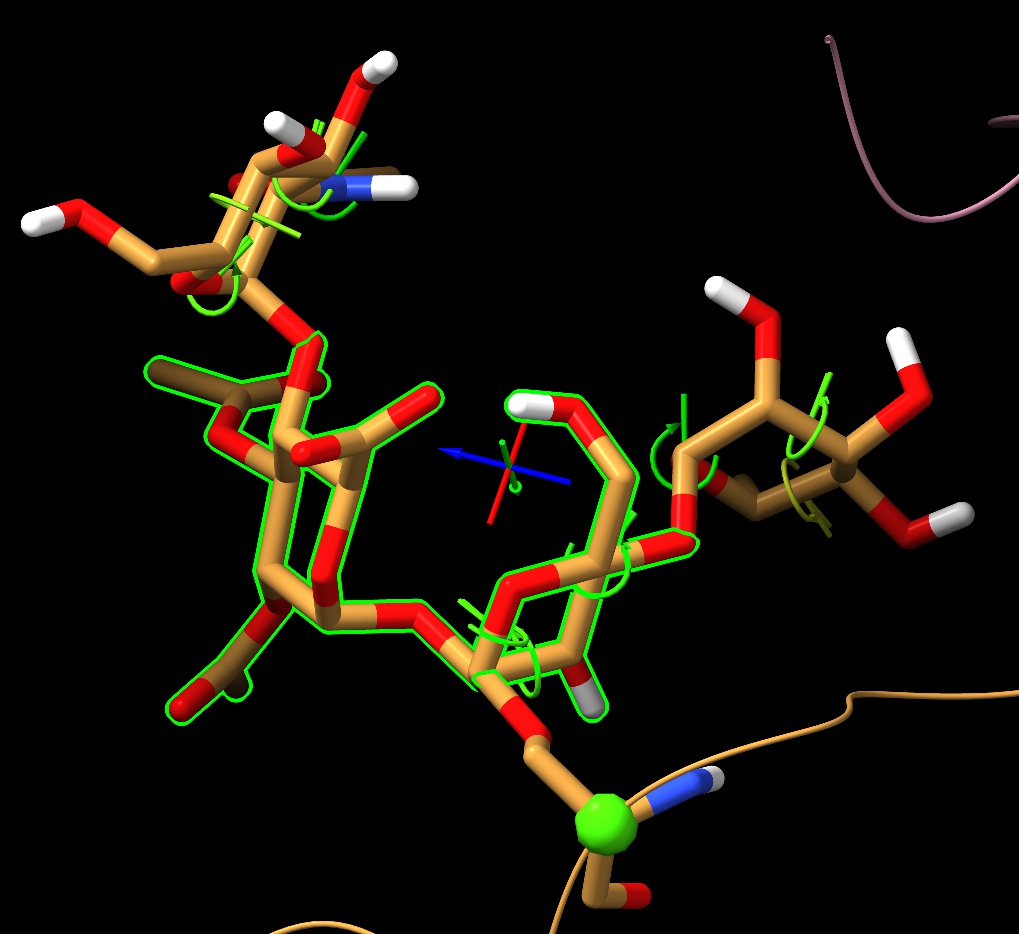{kind=link}
Note:
See TracTickets
for help on using tickets.
Added by email2trac