|
|
|
| ViewDock Interface (large image) |
Given the structures of ligand and receptor molecules, docking programs calculate possible binding modes. In virtual screening, small organic compounds (typically from a database of many thousands) are treated as possible ligands, and a target macromolecule is treated as the receptor.
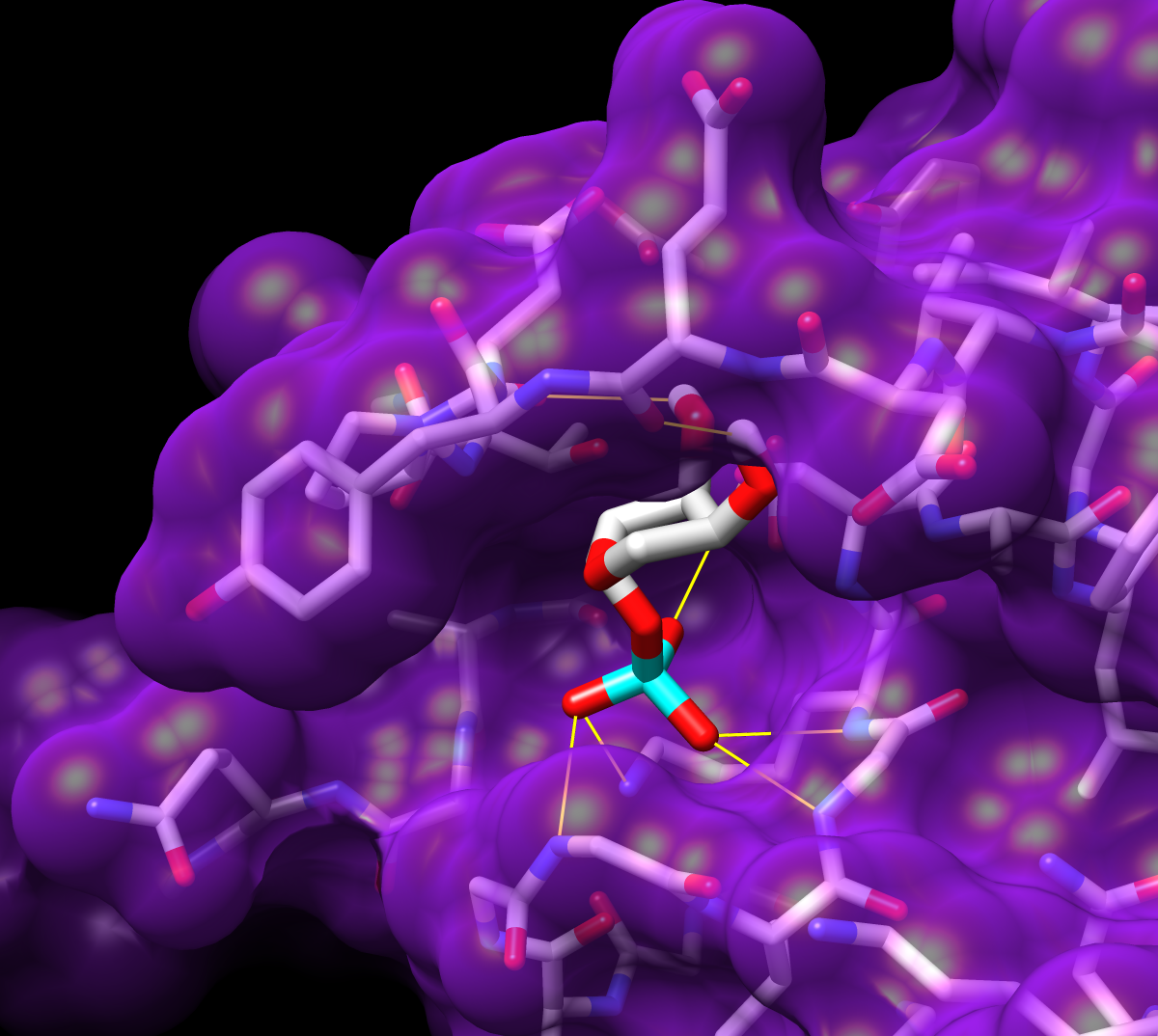
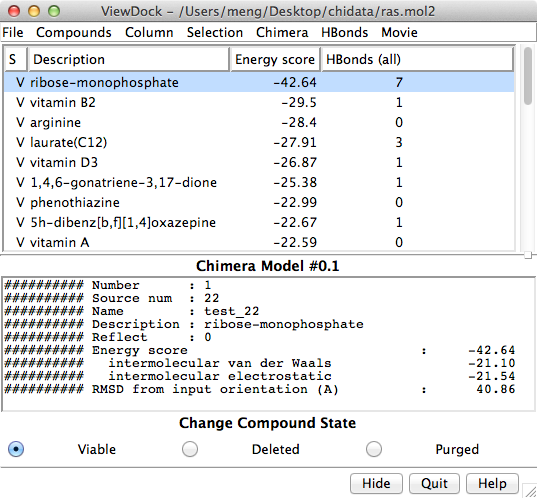
The Chimera tool ViewDock facilitates the interactive screening of compounds from the output of docking programs, including DOCK, AutoDock Vina, Gold, and others. The ViewDock interface (above, right) lists the docked molecules, and clicking on a line displays that molecule in the graphics window and shows information such as scores in the lower panel. The target structure in this example is H-Ras (PDB entry 121p), which is often mutated in malignant tumors in humans. The docked molecule is ribose monophosphate. Carbons are light gray, oxygens red, nitrogens blue, and phosphorus cyan. Hydrogens are not shown except for polar hydrogens (white) on the docked ligand. Potential hydrogen bonds are shown as yellow lines between donor and acceptor.
© 2004 The Regents of the University of California; all rights reserved.