

 home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search
home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search
home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search
home
overview
research
resources
outreach & training
outreach & training
visitors center
visitors center
search
search

John W. Sedat¹ and Tom Goddard²
¹
Department of Biochemistry and Biophysics
University of California, San Francisco
²
Resource for Biocomputing, Visualization, and Informatics
University of California, San Francisco
Within the higher eukaryotic chromosome, DNA is folded through DNA-protein interactions into multiple levels of organization. Together these yield greater than 10000:1 linear condensation of DNA into the fully compacted metaphase chromatid. From extensive biochemical characterizations as well as from a 2.8Å X-ray crystal structure, it is now reasonably clear how DNA is organized around the nucleosome core, the lowest level. However, as one progresses beyond the level of the nucleosome, the details of the higher orders of chromatin organization within chromosomes become increasingly uncertain; however, the importance of these higher-order organizations is clear. For example, there is strong evidence that the transcriptional repression of large chromatin domains is initiated and maintained through large-scale compaction. In order to more rigorously evaluate the structural variability of chromosomes within nuclei, and the relationship between chromosome structure and biological function, it is desirable to know the actual path of chromosomes within intact diploid nuclei.
In spite of considerable effort over the past decades, only recently has it been possible to determine the nature of even the simplest level of chromatin organization, the 30 nm fiber. The fundamental problem has been the extreme sensitivity of chromatin to isolation conditions, and the general lack of methods suitable for examining non-crystalline, non-symmetric, extremely complex and compact specimens in three-dimensions at moderately high resolutions (35-100Å). The combination of recent advances in sample preparation methodology (high pressure freezing, cryo-embedding, DNA-specific EM stains) and the refinement of both optical and electron microscopic tomography (EMT) methods for collecting three-dimensional data are crucial to the successful analysis of chromosome structure.
Two complementary efforts are underway to decipher nuclear and chromosome ultrastructure by optical and EMT methods. This includes the structural analysis of the Drosophila diploid anaphase chromosomes. These three-dimensional (3-D) chromosome structural studies have as their goal unperturbed chromosome states, hence a reference point for other structural studies such as in vitro preparations. All current EMT chromosome structure studies utilize High Pressure Freezing (HPF) technology, accomplished on the UC Berkeley-UCSF Shared HPF instrument, as a means to greatly preserve chromosome structure without tissue or chromosome isolation.
A major area of research is centered around the structure, largely unknown, of the diploid anaphase chromosome. Our goals are to extend our knowledge of the chromosome to three dimensions at both the optical and electron-microscopic resolution limits. The reconstruction, at high resolution, of specific anaphase chromosomes is underway. This direction is a continuation of ongoing work and has benefited enormously by complementary EMT efforts on other subcellular structures such as EMT reconstructions of the centrosome in which ~60Å resolution has been obtained. With some qualification, this level of resolution suggests that it will be possible to trace nucleosome structures through the higher-order chromosome organization, a long sought quest. Utilizing HPF technology means that in vivo preservation of chromosome structure is likely, thus solidifying the structural conclusions. Finally, we stress that preliminary analysis of the recent EMT reconstruction of the chromatids now allows one to follow substructures, strongly suggesting that these reconstructions will be interpretable. Diploid anaphase chromosomes are used as a diploid chromosome starting point as this is one of the few chromosome states that do not have attached homologues, sister chromatids or partial replication products.
Recent Advances
We continue to develop tools for image interpretation of chromosomal structure present in EMT data. We wrote and tested a comprehensive three-dimensional Fourier analysis and real space correlation software package (Chen, 1992) to study the chromosome reconstructions for the presence of repeating structural components and their sizes. The 3-D autocorrelation function spectrum, somewhat smoother than the Fourier analysis shows clear 110Å and 300Å peaks, plus others in the chromosome areas with no peaks in control nucleoplasm regions. Some of these peaks correspond to expected chromatin structures while other are novel suggesting higher order chromosome entities (or conceivable spacing dimensions between structures). We wrote extensive software to generate filters that bring out visually the structures likely present. We tried Fourier filters of many kinds: matched filters, wavelet analysis, and edge detection schemes of various kinds, always using as controls the chromosome-free nucleoplasm regions. While there were abundant hints in the filtered images of what appeared to be clear helical structures in many places on the chromosomes, the resulting images were not unambiguous. We continue to search for optimum filters using new approaches, however.In the past years, several new directions have been undertaken. First, new optical microscopy (structured illumination, SI -resolution ≤100nm) in an easy-to-use format has been developed. Second, we have applied this technology to anaphase chromatids with intriguing substructure now visible. Third, we developed the procedures to lift off the sections, after SI optical microscopy, restain and then carry out EM tomography. This means we have both optical microscopy (OM) and EMT data for the same sample. Fourth, we have computationally refined the EMT of the anaphase chromatids-fixing small misalignments, intensity problems, missing data at high angles (missing wedge problem)-using software developed by Dr. David Agard and ourselves (TAPIR).
What results is the best chromosome data we have seen in 30 years of working on chromosome structure. We now can see extensive multiple levels of higher order chromosome structure and we are intensively interpreting this data.
Simultaneously with the advances on chromosome structure, Chimera grey-level modeling capability has progressed. We now find that we can model - find and model the densities - in stereo with careful control of all aspects of the 3-dimensional images. We now have grey level modeling capability (and flexibility) approximately equal to what protein x-ray crystallography has, but for large-scale cellular structure. This new Chimera capability is essential and we currently are full time, with several people, modeling and interpreting the optical and EMT anaphase chromatids. This analysis effort will progress and be extended in a proposed new Sedat lab grant as a major focus.
Barcode Labeling of Chromosomes Complementary to the EMT methods above, studies were underway to utilize fluorescence in situ hybridization (FISH) techniques in combination with 3-D optical microscopy. While this approach can detect single copy loci, the limited number of spectrally distinguishable fluorophores severely limits our ability to sample the position of the chromosome fiber at many loci simultaneously. To circumvent this limitation, graduate student Michael Lowenstein has modified existing FISH techniques such that each fluorophore can be used to localize many discrete loci. This method involves labeling a set of ordered probes with one of three or more fluorophores such that a color "barcode" is embedded along the linear chromosome. After co-hybridizing the entire probe set and then imaging samples, a data set is generated for each nucleus with multiple fluorescence spots in each wavelength. Distinguishing among identically labeled probes (and thus tracing the chromosome path) is possible because the barcode and the intrinsic distance geometrical constraints of the linear chromosome constrain any plausible global solution.
Using this technique two types of useful information can be obtained. First, given the distance between many adjacent loci we can evaluate local variations in compaction, which are hypothesized to correlate with changes in expression. Second, by measuring all inter-probe distances (including between homologs), and the distance of all probes to the nuclear envelope, we can identify features that define higher order nuclear organization. The long-term objective of these experiments is to understand how higher order chromosome structure changes as a function of gene expression, the cell cycle, and cellular development.
We have recently used a tri-color, 13-probe barcode (see below) to evaluate the structure of Drosophila melanogaster chromosome 2L in embryonic nuclei. In addition to proving the feasibility of bar-coded FISH, we provide previously unavailable information on the in vivo configuration of interphase chromosomes. The key challenge now is to interpret the 3-D structures we have observed. Toward this goal, we have worked with Tom Goddard in the RBVI to develop software visualization tools for 3-D microscopy data. A Chimera capability to interactively trace chromosome paths was developed, similarities of traces were analyzed using alignments, and traces were compared to paths generated by stochastic models. This work, which benefited greatly from Chimera, is now published (Lowenstein, 2004), with Tom Goddard a co-author.
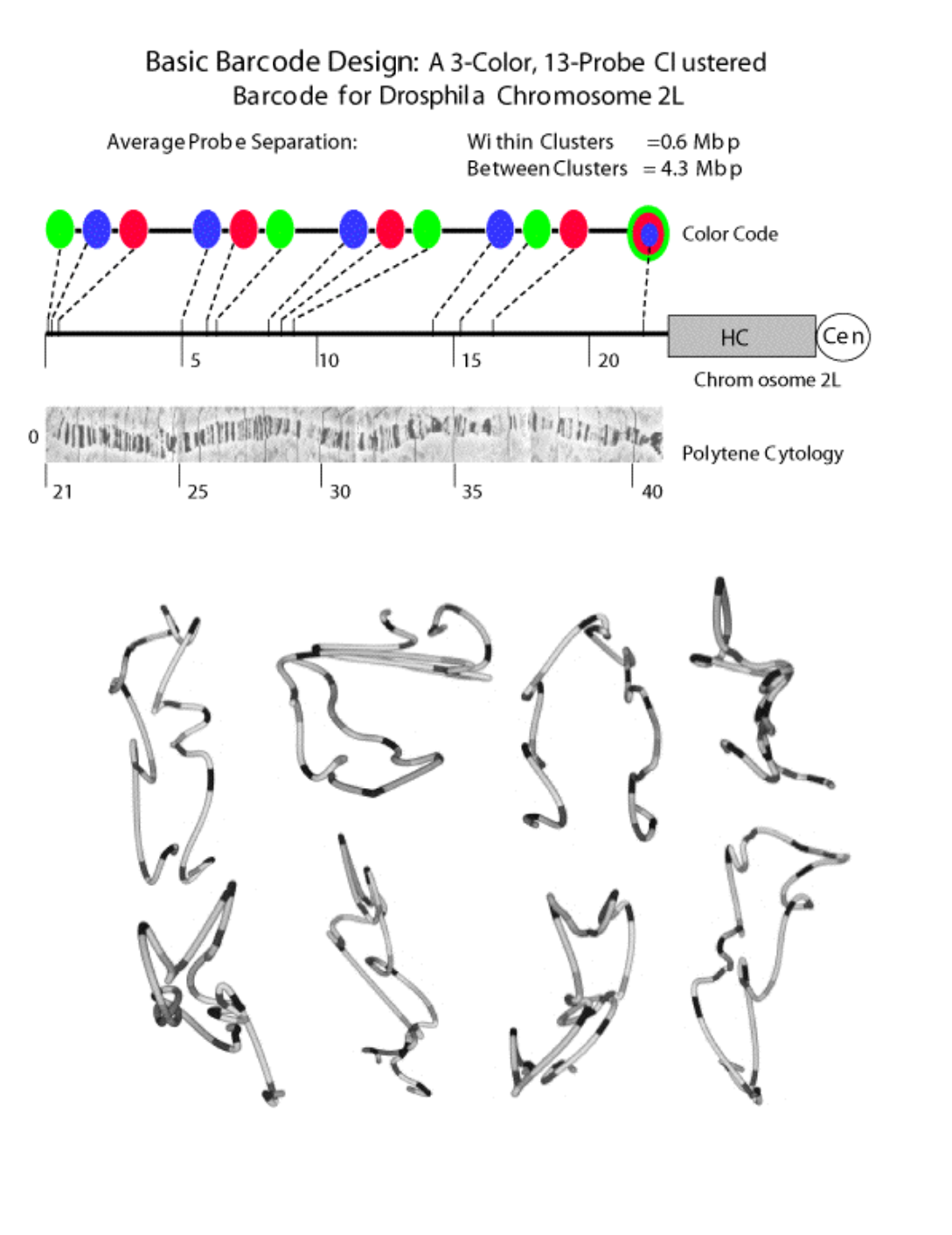
Figure caption: Top: A set of probes is chosen which map sequentially along the target chromosome. Color assignments and probe spacing are chosen to minimize assignment errors during the decoding (tracing) step. The barcode shown is for Drosophila chromosome 2L. It uses 3 fluorphores (FITC, Rhod and Cy5 directly conjugated to dUTPs). The code has 13 probes spaced in 4 clusters of 3 (with each fluorphor used only once), with an anchor point provided by a triple labeled His probe. With this barcode a total of 26 unique loci can be discriminated in diploid nuclei. The probes are from the sequenced Drosophila P1 genomic library and each contain about 80kbp of sequence. Bottom: Connectivity is determined by globally minimizing the solution path length constrained by additional contextual information (polarized chromosome orientation, probe signal intensity). Once the path is determined, an interpolated tube model is drawn which can be rotated and further evaluated by visual inspection within the Chimera modeling environment.
High Spatial and Temporal Resolution Optical Microscopy
We continue to push the development of the instrumentation because it has proven to be so useful (and essential) for the chromosome structure information. The novel OMX microscope designed and built by our lab started collecting data in 2004. It is capable of both very fast live imaging and easy-to-use structured illumination image acquisition (resolution ≤100 nm, beyond the diffraction limit) at four simultaneous wave lengths and is now fully functional with much use by our group, as well as other groups (really around the world; it is amazing how spectacular the data is). We also continue to develop, as a major rebuild with new capabilities, the interferometric optics microscope (which provide ≤100 nm x,y and ≤80-90 nm in z resolution), as a project of a graduate student with several papers in preparation.An interesting feature of OMX is the very fast 3-D imaging capability. What results is a very densely sampled time domain, something we have not had before. There is currently a large effort to interpret the time data in new ways. It should be possible to correlate, in several ways, the time points to see which image feature coherently correlate between the time points with noise possibly not correlating. A large number of filters (e.g. median, Fourier, and wavelet) might also improve our ability to interpret this data.
Future Chimera Projects
Three projects in our lab could benefit from interactive analysis of density maps using Chimera.First is the time series data analysis, 4-D data from OMX, tracking features in that data. Chimera could be useful in visualizing substructure and particle tracking. What one envisions is that particle tracking or future worm tracking can be overlaid in Chimera on the density data. You would be able to see one time frame in stereo and see a trajectory including past and future positions of particles or worms displayed as a model such as a smooth time-trace curve, or for a worm, a series of successive worms. You would be able to control exactly how much of these trajectories in the past and future are displayed. This tool would visually assess results from tracking methods. One is also interested in developing a motion tracking approach that calculates how to minimally move the mass in the density map from one frame to the next. This would give a vector flow that would allow tracking any particles or features. Given that flow one could by hand color a particular chromosome in a mitotic cell red, one blue, one yellow, then see through mitosis how those chromosomes move. In reality tracking is likely to break down at points where the chromosomes cannot be distinguished at available resolutions. Of course, we must explore the limits of this analysis method.
The second problem our lab is studying where we believe Chimera will be relevant is superposition of EMT and OM data of the same sample. The optical and EMT maps showed striking differences where individual chromosomes did not align, and density gradients perhaps reflecting uneven staining were apparent. In essence, the two types of data provided a crosscheck on the correctness of the density maps. The study of variables that effect EMT and OM data quality is crucial to producing data that will reveal chromosome biology. Here one sees Chimera helping in a couple ways. One would want to add alignment optimization using density map correlation. It would be used to align OM and EMT data. As the case above illustrates, not all parts will align. By locally aligning features that seem to have moved, exact quantification of how much a chromosome rotated, translated and shrunk could be made. Once the OM and EMT data are aligned, it would be interesting to filter the EMT data to artificially lower the resolution to that comparable to the OM map and then look at differences. The differing regions can then be inspected in the full resolution EMT data. This involves some new interactive filtering (Gaussian convolution), which is already a planned addition to Chimera, and new difference map analysis tools, for example.
The third area is analysis of EM tomography of anaphase chromosomes. An initial step to analyze this data is to hand trace structures by looking at the EMT maps filtered to a range of different resolutions. If common substructures are observed, it would make sense to computationally enhance those features, or find all occurrences in the map, and run a statistical analysis to confirm that the features are real. We often see helical structure, and hand-tracing some dozens to hundreds of substructures would reinforce our belief in these observations. Then it would make sense to fit a left-handed or right-handed bent arc of sizes suggested by the hand-traces throughout several data sets to see if that substructure is statistically significant. A big question regarding the EMT anaphase analysis is whether some pre-filtering to enhance visibility of the substructures is essential to be able to hand trace a feature in the first place.
References:
- H. Chen, W.K. Clyborne, J.W. Sedat, and D.A. Agard, "PRIISM: an integrated system for display and analysis of 3-D microscope images," Proc. SPIE, 1660:784-790, 1992.
- M.G. Lowenstein, T.D. Goddard, J.W. Sedat, "Long-range interphase chromosome organization in Drosophila: a study using color barcoded fluorescence in situ hybridization and structural clustering analysis," Mol. Biol. Cell, 15(12):5678-92, 2004.
Laboratory Overview | Research | Outreach & Training | Available Resources | Visitors Center | Search